
2010年8月2日
林 憲一
2009年11月下旬から、ロンドンにあるEuropean Medicines Agency(以下Agencyという)にMHLW/PMDAのLiaison Officialとして派遣され、長官オフィス(Office of the Executive Director)に籍を置くこととなりました。この機会に、Agencyに関する情報や話題について、Canary Wharf便りと題して報告したいと思います。
なお、このCanary Wharf便りは、Agencyに派遣されている林がAgencyに関する情報を個人の立場でまとめたもので、Agencyあるいは派遣元である厚生労働省(MHLW)、医薬品医療機器総合機構(PMDA)の見解等を示すものではないことにご留意下さい。
ヒト用医薬品委員会(CHMP)
6月21-24日に開催されたCHMP(Committee for Human Medicinal Products)でChair、Vice-Chairの改選があり、Dr Eric Abadie(フランス)がChairに、Dr Tomas Salmonson(スウェーデン)がVice-Chairにそれぞれ再選されました。2期目のChair、Vice-Chairを3年間務めることとされています。
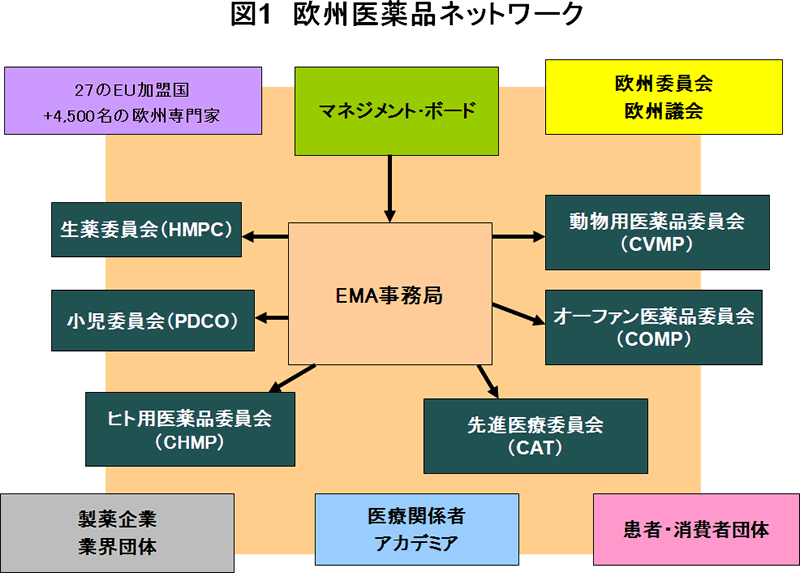
これまでにも何度かご紹介しましたが、CHMPをはじめとするEMAの科学委員会(Scientific Committee)は、27のEU加盟国、EEA-EFTA諸国等のメンバーから構成され、さらに各国の4,500名を超える専門家のネットワーク(図1)にも支えられて、Agencyがその機能を果たしていく上で重要な役割を果たしています。
{kind=link}
特にCHMPについては、規則(EC)No 726/2004のヒト用医薬品のMarketing Authorisation(MA)審査の手続きを定めた第5条以下で、中央審査手続きに従って提出されたヒト用医薬品のMA申請資料の受入れ、承認、承認事項の一部変更、承認の一時停止または取消しに関するあらゆる疑問に対しAgencyとしての見解を用意する責務を負うことが規定されており(Agencyのtaskを定めた第56条にも関連する規定が見られる)、医薬品の品質・安全性・有効性の評価において中心的な役割を担っています。この目的のため、CHMPは中央審査手続きに従い共同体全域でのMAを求める医薬品に対して最初の評価を実施するほか、承認後のメンテナンスに係る評価(承認事項の一部変更、ファーマコビジランス等)も担当しています。
CHMPのメンバー構成は、27の各加盟国から指名されたメンバー1名とその代理、EEA-EFTA諸国のノルウェー、アイスランドから各1名とその代理、5名の選任メンバーです。選任メンバーとは、生物由来製品の品質・安全性、薬剤疫学、生物統計など特に高度な専門性が求められる領域についてCHMPが追加で専門家を選任するシステムにより選ばれた人たちです。
CHMPの会議は原則として8月を除き毎月開催されます。会議の日程は5年先まで決められており、毎月4日間、毎日午前8時半から午後7時過ぎまで相当数に上る議題を次々にこなしていきます。タイトなスケジュールのため当日出席できない専門家が、関係する議題に関して電話で討議に参加するシーンもしばしば見られます。また、緊急の案件が生じた場合には臨時の会議が開かれることもあります。
EU加盟国間の相互承認や非中央審査手続きでは、CHMPが仲裁者として特定の医薬品に関して加盟国間で意見の相違がある場合に調整を行ったり(arbitration procedure)、公衆衛生上の懸念その他共同体の利益が脅かされるような問題が起きた際に照会に対して意見を述べたりする(Community referral procedure)機能があります。
CHMPによって行われる評価は、純粋に科学的なクライテリアに基づいて行われ、その医薬品が品質、安全性、有効性について必要とされる要件を満たしているか否かを、EUの法令、特に指令2001/83/ECの規定に従って確認していきます。このプロセスにより医薬品が市場に出たときに患者/ 使用者にとって望ましい(ポジティブな)リスク/ベネフィット・バランスを有しているかどうかの確認が行われます。指令2001/83/ECでは、CHMPは見解を出す前にMA申請者またはMA保持者に書面または口頭で意見を述べる機会を与えるとされていることから、文書回答に加えてCHMPでのoral explanation(いわゆるプレゼンテーション)を希望する製薬企業もあります。
CHMPの見解は基本的にコンセンサスにより決定されますが、専門家の意見が分かれてコンセンサスが得られない場合には、投票による決定が行われます。投票は賛成、反対のいずれかで棄権は認められていません。意見の採択は、投票資格を持つメンバーの過半数の賛成で決定され、対立する意見があれば記録に残されます。ノルウェーとアイスランドは投票はできるものの他の加盟国とは別に扱われ、委員会決定のための票数にはカウントされません。
医薬品が承認された後は、EU各国の医薬品規制当局によるネットワークが医療関係者や医薬品を販売している製薬企業と連携して安全性モニタリングを行いますが、CHMPはこのEUのファーマコビジランス活動でも重要な役目を担っており、各国のファーマコビジランス・システムを通じて集積された安全性情報をモニターすることにより、必要な場合には欧州委員会に承認内容の変更や販売の一時停止またはMAの取消しなどの勧告を行うこととされています。安全性の理由から緊急にMAの内容を変更する必要があるときは、urgent safety restriction(USR)を出して変更の内容およびどんな場合に使ってよいか等の情報を医療関係者に知らせます。
CHMPでは、中央審査手続きによりMAを与えられた全ての医薬品に対してEuropean Public Assessment Report(EPAR=CHMPが行った評価の概要を記した報告書。一般の人にも分かりやすい言葉で書かれ、原則として全てのEU公用語に翻訳される。EMAウェッブサイトに掲載されている)を公表し、委員会として承認するに至った科学的根拠を明らかにしています。またsummary product characteristics(SmPC=医薬品の品目概要。MAにおける承認事項とされており、医療関係者や患者への情報提供、製品の表示、広告などはすべてこのSmPCの記載に基づいて行わなければならない)、ラベリング、パッケージングの要件、評価プロセスの各段階における手続きの詳細に関する情報なども公表されています。
この他にCHMPでは、科学的アドバイスを含めた製薬企業の新医薬品の研究開発の支援、企業が参照するための科学的または規制に関するガイドライン文書の作成、国際的なパートナーと協力して医薬品規制の調和を進める活動等を行っています。
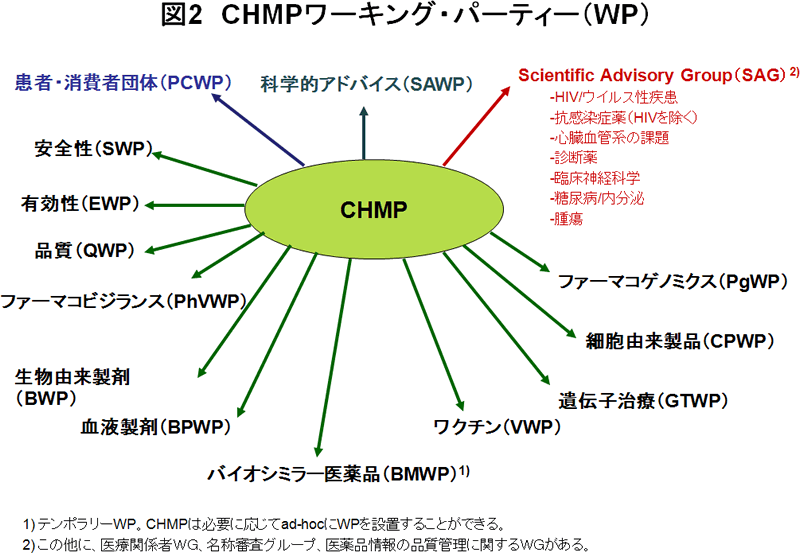
ヒト用医薬品開発/評価ユニットの説明(第2回Canary Wharf 便りの1.参照)のところでも少し触れましたが、CHMPにはワーキング・パーティー(WP)等の下部委員会が置かれています(図2)。これらのWPはEMAが保有する欧州専門家リストから選ばれた特定の分野の専門知識を有するメンバーから構成されています。CHMPは科学的課題について各分野のWPの意見を聴いたり、MA申請に関する事項やガイドライン案の作成・改正作業の検討などをWPに行わせたりします。そのためCHMPでは個々の医薬品の審議とは別に、各WPから活動のアップデートやガイドライン案の検討状況等の報告を行う時間が設けられています。
{kind=link}
WPには安全性WP、有効性WP、CHMP/CVMP合同品質WP、ファーマコビジランスWP、生物由来製剤WP、血液製剤WP、ワクチンWP、遺伝子治療WP、細胞由来製品WP、ファーマコゲノミクスWP、科学的アドバイスWP、患者・消費者団体WPがあります。また臨時の作業が必要な場合には、CHMPはテンポラリーWPを設置して作業を行わせることができます。このWPでは特定のトピックに対する提案やCHMPで提議された疑問への回答案の作成、当該WPが専門性を有する分野のガイドライン案の作成や改訂作業が行われます。現在設置されているテンポラリーWPとしては、バイオシミラー医薬品WPがあります。
一方、WPとは別にCHMPの下にはScientific Advisory Groupという組織が置かれています。SAGは特定の医薬品や治療法の評価について助言を行う組織として設置されているものです。SAGもCHMPまたはEMAが専門性に応じてノミネーションした中から選ばれた欧州専門家により構成されています。
SAGには、HIV/ウイルス性疾患に関するSAG、抗感染症薬に関するSAG、心臓血管系の課題に関するSAG、診断薬に関するSAG、臨床神経科学に関するSAG、糖尿病/内分泌に関するSAG、腫瘍に関するSAGがあります。この他にSAG関連グループとしてCHMPにより、医療関係者WG、名称審査グループ、医薬品情報の品質管理に関するWGが置かれています。CHMPはこれらのグループにMA、申請、ガイドラインのドラフティングや改訂等に関し意見を聴いています。さらにGMP、GCP、GLP査察業務グループによってもCHMPはサポートされています。
CHMPによる科学的評価の作業はAgency内部のピア・レビュー・システムの対象とされ、委員会の出す見解の正確さや妥当性を担保するための努力がなされており、CHMPの各プロセスと記録の運営・管理がAgencyの総合マネジメント・システム(IQM)を通じて行われています。
EMAに対する評価の結果に関するカンファレンス
2010年6月30日に欧州委員会(European Commission)とEMAの共催で、2009年にCommissionがErnst & Youngというコンサルタント会社に委託し2010年4月に公表されたEMAおよびEMAとEU各国とのネットワーク(図1)の効率性や効果の評価の結果に関するカンファレンスがEMA内で開催されました。会議のプレスリリースが以下のリンクに掲載されています。
European Medicines Agency and European Commission start reflection process on way forward for the Agency and the network
カンファレンスには、EMA、Commission、EU各国当局の他、産業界、患者・消費者団体の代表、ジャーナリストら約150名の関係者が参加し、評価の結果とAgencyの将来の課題解決に向けてそれをどう生かしていけばよいかが議論されました。
カンファレンスでは、Agencyの科学委員会の構造とAgencyの調整機能(Architecture of the European Medicines Agency's Scientific Forum)(特にCHMP等の責務の増大に伴い委員会間の複雑なinteractionやworkloadが増加している問題)、European Medicines Network(EUネットワークを通じて域内の専門家を長期的に確保するための方策)、動物用医薬品に係る課題、Agencyが活動に必要な原資を長期間安定的に確保するための課題(Fee System of the European Medicines Agency)、Road Map(2015年までのロード・マップに対して寄せられたパブリック・コメント)等の紹介が行われました。
それに対し、ますます国際化する医薬品産業にAgencyはどうすれば効果的に対応できるか、先進医療やパーソナライズド・メディシンにふさわしい体制がとられているか、患者・消費者団体からの参加・透明性向上の要請に十分応えられているか、といった指摘が聞かれました。
Ernst & Youngの評価報告書はCommissionのサイトに掲載されています。
Evaluation of the European Medicines Agency – Final report – January 2010
報告書では、これまでEMAが人や動物の健康保護に有効な機能を果たすことに成果を挙げ、欧州のみならず国際的にも高い評価を獲得してきたことが再確認される一方、CHMP等におけるworkloadの増加などの課題があることを指摘し、しかしEMAが組織の持続可能性を維持しながら自己変革を遂げていくなら、今後直面するであろう科学的(emerging new technology)、社会的(growing needs for geriatric products)、組織的(臨床試験をscope of EMA competenceの中に含めるべきであるとする動き等)課題にも適応していけるだろう、と結ばれています。
Lonngren長官からは「EMAの成功は各国当局、欧州委員会、Agencyの三者の協力の賜物である。このネットワークモデルは15年間よく機能してきた。しかし、Agencyを取り巻く科学的、政治的、社会的状況が変化し、Agencyの構造をもう一度よく見直し、目的に合致したものにする必要がある。この内省のプロセスを関係者にもオープンにし、可能な限り広い範囲のコンセンサスを得て将来のAgencyの方向性を決めていきたい」とコメントがありました。
今後、EMAは欧州委員会と協力して合同報告書を作成し、カンファレンスで示された意見や提案にどのように取り組んでいくか明らかにするということです。
EMAの新しいウェブサイト
EMAのコーポレート・ウェブサイトが、7月15日付けで全面リニューアルされました(第1回Canary Wharf便りの2.参照)。
European Medicines Agency launches new website
EMAのウェブサイトには月平均50万回のビジットがあり、サイトを閲覧する患者、医療関係者、規制関係者等、医薬品の規制や安全性に関心のある人たちの重要な情報源となっています。
今回のリニューアルでは、デザインが一新されて直感的な操作がしやすくなっただけでなく、ヒト用、動物用医薬品、生薬製剤のクイック検索機能、Agency文書のオンライン・ライブラリー、オンライン上で情報が公表されるとその情報が直ちに届くRSS feedsなどの新たな内容・機能も付加されて、EMAに関する情報の利便性が高まるとともに、活動の透明性の向上も併せて図られています。
Partners & Networksの項にはMHLW/PMDAの英文サイトへのリンクも張られています。
Agencyでは今後もマルチメディアの利用や検索機能の改良など、引き続きウェブサイトの改善に力をいれていくそうです。