
Nobuo UEMURA 10/27/2010
Foreword
In mid-February I moved to the United States Pharmacopeia (USP) secretariat office as a visiting scientist of USP General Chapter team from Japan and a liaison official of MHLW/PMDA ("Ministry of Health, Labour and Welfare" and "Pharmaceuticals and Medical Devices Agency").
This is the fourth report on the USP and FDA issues. However this report is my personal views and opinions, and it does not necessarily represent the formal position of MHLW, PMDA, FDA and USP.
1. USP Reference Standards
USP Reference Standards are integral components of monographs and other documentary standards established by USP to help ensure the identity, strength, quality and purity of medicines. These Standards provide the necessary reference values or characteristics for use in conducting the assays and tests in the official documentary standards; United States Pharmacopeia-National Formulary(USP_NF).
USP official Reference Standards are highly characterized specimens of drug substances, excipients, impurities, degradation products, dietary supplements, compendia reagents and performance calibrators.
USP Reference Standards are available for methods and procedures specified in all of the USP compendia including USP-NF, the Food Chemicals Codex, the Dietary Supplements Compendium, and the Pharmacists' Pharmacopeia.
USP offers more than 2,500 Reference Standards (as of May 2010) for pharmaceuticals, excipients and dietary supplements, and these standards are used in more than 130 countries around the world.
There are four main types of USP Reference Standards: 1) Standards with Quantitative Application (Assays and Limit tests), 2) Standards with only Qualitative Application (Identification tests, Elution markers, System Suitability tests), 3) Performance Verification Standards (Dissolution standards, Melting point standards, Particle count Reference Standards), 4) Standards that define a unit of activity (Complex antibiotics, Endotoxins, Enzymes etc.).
USP laboratories and Reference Standard Production Facilities provide these Reference Standards through a collaborative testing process that involves at least three independent laboratories. In addition to USP's laboratories in Rockville, China, India, and Brazil, USP collaborates with the FDA, Health Canada, UK NIBSC, Australia TGA, China SIFDC and NICPBP. For some biological Reference Standards where potency is assigned, USP assays its Reference Standards against WHO International Standards.
USP Reference Standards are available by order to USP or through the authorized distributors in outside US countries.
USP Reference Standards
Reprinted with permission from the United States Pharmacopeial Convention, 12601 Twinbrook Parkway, Rockville, MD 20852, October 2010
2. Pending Monographs
USP provides a Web-based publication of Monographs for articles (e.g. drug substances, excipients and dosage forms) from 2007 based on information in FDA generic (or OTC) applications and filings awaiting FDA approval. These monographs, called as "Pending Monographs", are not part of an official compendium of USP or NF and not legally enforceable.
The purpose of the Pending Monographs is to have an official USP or NF monograph ready as soon as possible after FDA grants final product approval. To this end, the Pending Monograph development begins before FDA's approval process is complete, resulting in an official USP or NF monograph more rapidly than would be possible if monograph development started only after final FDA approval.
In accordance with the guideline, requests for pending monograph will be accepted only from sponsors who have filled or intend to file with FDA an Abbreviated New Drug Application (ANDA), a Biosimilar or Interchangeable Biologics License Application (Public Health Service act 351(k) BLA), or who have submitted a Drug Master File (DMF) for an article to FDA, or whose substance is or will be the subject to the FDA OTC drug monograph being amended.
As new or revised Pending Monographs in draft become available, they will be posted on the USP Web site for requesting public comments. Following review of comments and approval by the relevant USP Expert Committee, the authorized Pending Monographs will be posted to this USP Web site.
Once FDA approval is granted, USP then work with the sponsor to move that monograph into official USP or NF status.
As of September 2010, 43 authorized and 18 draft Pending Monographs are listed before FDA approval, and 12 Monographs have either advanced to or are in the process of advancing to official status.
3. USP Non-US Monograph
From 2007 USP provides voluntary public documentary standards (Monographs) for drug products and their ingredients legally marketed in countries outside the United States if these drugs are intended to treat neglected diseases. The purpose of these monographs is to support testing by manufacturers, users, government control laboratories etc. Where appropriate, USP will provide reference materials to support testing of the article against the Non-US Monograph requirements.
The Non-US Monograph is not part of an official compendium of the US (USP or NF), but manufacturers of drug substances or drug products may use the designation "S-USP" on certificates of analysis or on container labels, respectively, to indicate compliance to these documentary standards for their drug products or drug substances.
The Non-US Monograph is available only on Web site, and as of September 2010, around 200 drugs or their combinations are listed for HIV/AIDS, AIDS-associated opportunistic infections and other neglected diseases.
Example of neglected diseases;
African trypanosomiasis, Hemorrhagic fever with renal syndrome due to Hantavirus, Lassa fever, Leishmaniasis, Malaria, Schistosomiasis, Tuberculosis, Chagas disease, Leprosy, Lymphatic filariasis, Onchocerciasis, etc.
4. Funding by Continuing Resolution
The 2010 midterm election is scheduled in November in this year, and the appropriation bills were not completed by the end of September.
The Congress authorized the Continuing Resolution in late September. The temporary funding for the administration's activities which was necessary to maintain the citizen's life and security, and was requested for FY2011 budget at FY 2010 enacted levels, are approved to allow continued government operations from October 1 through December 3.
FDA is continuing their consecutive activities which are listed in the FY2011 budget request.
5. Performance Report of Federal Agencies
The Government Performance and Results Act 0f 1993 (GPRA), (Pub. L. No. 103-62), requires Federal agencies to make and submit Strategic Plans, Performance Budgets/Annual Performance Plans, and Annual Performance Reports.
The annual performance report, required by GPRA, provides information on the agency's actual performance and progress in achieving the goals in its strategic plan and performance budget.
FDA's performance report in the Online Performance Appendix is one of several documents that fulfill the Department of Health and Human Services' (HHS) performance planning and reporting requirements.
The FY 2011 Congressional Justifications and accompanying Online Performance Appendices contain the updated FY 2009 Annual Performance Report and FY 2011 Annual Performance Plan.
The Agency Performance Reports and Financial Reports are sent to the President, Congress and the Director of Office of Management and Budget(OMB).
6. CDER/CBER Performance Report
Main Performance Goals of CDER and CBER from FY 2008 to FY 2012 are as follows;
1) Human Drug Performance Detail
Long Term Objective : Improve the medical product review process to increase the predictability and transparency of decisions using the best available science.
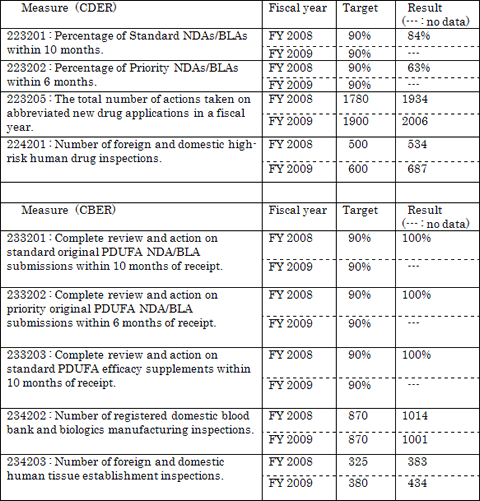
223201 : Percentage of Standard NDAs/BLAs within 10 months.
223202 : Percentage of Priority NDAs/BLAs within 6 months.
223205 : The total number of actions taken on abbreviated new drug applications in a fiscal year.
223207 : Reduction in FDA approval time for the fastest 50 percent of standard New Molecular Entities/Biologics Licensing Applications approved for CDER and CBER, using the 3-year submission cohort for FY 2005-2007.
223208 : Reduction in FDA time to approval or tentative approval for the fastest 70 percent of original generic drug applications approved or tentatively approved of those submitted using the 3-year submission cohort for FY 2005-2007.
Long Term Objective : Improve information systems for problem detection and public communication about product safety.
222303 : Improve the safe use of drugs by patients and health care providers by reviewing safety labeling changes required under FDAAA within the timeframes established by FDAAA.
222201 : The Unit Cost associated with turning a submitted Adverse Event Report into a verified record in the database.
222202 : The percent of manufacturer submitted expedited adverse event reports received electronically compared to all expedited adverse event reports received from industry.
Long Term Objective : Detect safety problems earlier and better target interventions to prevent harm to consumers.
224201 : Number of foreign and domestic high-risk human drug inspections.
222302 : Percentage of television advertisements requiring submission reviewed within 45 days.
2) Biologics Performance Detail
Long Term Objective : Increase the number of safe and effective new medical products avail to patients
233201 : Complete review and action on standard original PDUFA NDA/BLA submissions within 10 months of receipt.
233202 : Complete review and action on priority original PDUFA NDA/BLA submissions within 6 months of receipt.
233203 : Complete review and action on standard PDUFA efficacy supplements within 10 months of receipt.
233205 : Complete review and action on complete blood bank and source plasma BLA submissions within 12 months after submission date.
233206 : Complete review and action on complete blood bank and source plasma BLA supplements within 12 months after submission date.
Long Term Objective : Prevent safety problems by modernizing science-based standards and tools to ensure high-quality manufacturing, processing, and distribution.
234101 : Increase manufacturing diversity and capacity for pandemic influenza vaccine production.
Long Term Objective : Detect safety problems earlier and better target interventions to prevent harm to consumers.
234202 : Number of registered domestic blood bank and biologics manufacturing inspections.
234203 : Number of foreign and domestic human tissue establishment inspections.
The table below summarizes FDA's review and other performance for FY 2008 and FY 2009 to meeting PDUFA performance goals. (As of September 30, 2009)
7. CDRH Performance Report
Review Performance Goals of CDRH from FY 2008 to FY 2012 are as follows;
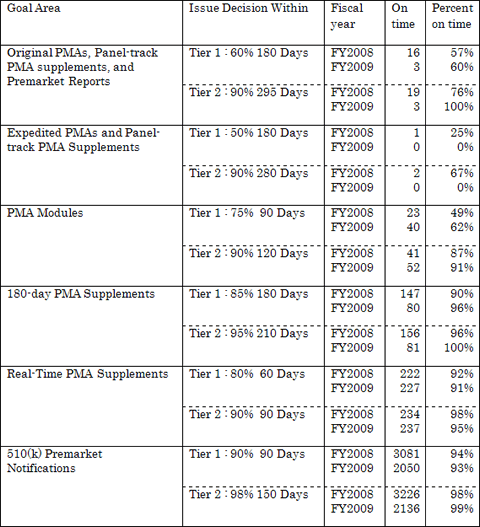
1) Original Premarket approval (PMA), Panel-Track PMA Supplement, and Premarket Report Submissions
FDA will issue a decision for 60% of non-expedited filed submissions within 180 days, and for 90% within 295 days.
2) Expedited Original Premarket approval (PMA) and Panel-Track PMA Supplement Submissions
FDA will issue a decision for 50% of expedited filed submissions within 180 days, and for 90% within 280 days.
3) PMA Modules
FDA will take action on 75% of PMA modules within 90 days, and on 90% within 120 days.
4) 180-Day PMA Supplements
FDA will issue a decision for 85% of 180-day PMA supplements within 180 days, and for 95% within 210 days.
5) Real-Time PMA Supplements
FDA will issue a decision for 80% of real-time PMA supplements within 60 days, and for 90% within 90 days.
6) 510(k) Submissions
FDA will issue a decision for 90% of 510(k)s within 90 days, and for 98% within 150 days.
7) Maintenance of Current Performance
The agency will, at a minimum, maintain current review performance in review areas such as IDEs and 30-day Notices where specific quantitative goals have not been established.
In addition, the following items of goals and procedures are summarized under the medical device user fee program in the Medical Device User Fee amendments of 2007;
- Interactive review
- Meetings
- Quarterly Performance Reports
- New Commitments
- Guidance document Development
- Reviewer Training
The table below summarizes FDA's review performance for FY 2008 and FY 2009 submissions subject to meeting MDUFA II performance goals. (As of September 30, 2009)
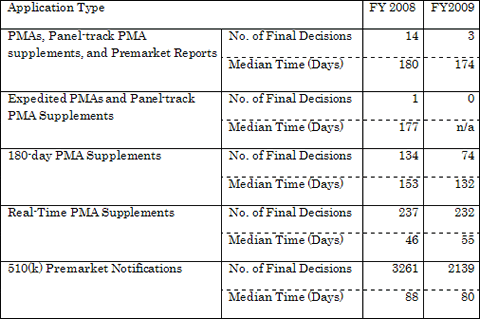
The table below presents the total median review times to final decisions for each application type.
8. Cross-agency Goal and Budget Submission for FY 2012.
Obama Administration has launched numerous cross-agency collaborations to promote efforts among agencies to work effectively together to achieve Presidential priorities.
OMB encourages agencies to consult with each other during the budget planning process so that resources are allocated to maximize their impact and avoid inappropriate duplication.
OMB will formalize efforts that working groups have been exploring in following several areas to coordinate FY 2012 Budget submissions among relevant agencies.
Science, Technology, Engineering and Mathematics education;
Large ecosystem restoration;
Climate science;
Climate technology;
Clean energy;
Nanotechnology;
Computing research;
Homelessness reduction;
Place-based policies;
and Obesity reduction
This effort has started by the OMB's announcement in June 2010.