
Nobuo UEMURA 2/28/2011
Foreword
In mid-February, 2010, I moved to the United States Pharmacopeia (USP) secretariat office as a visiting scientist of USP General Chapter team from Japan and a liaison official of MHLW/PMDA ("Ministry of Health, Labour and Welfare" and "Pharmaceuticals and Medical Devices Agency").
This is the sixth report on the USP and FDA issues. However this report is my personal views and opinions, and it does not necessarily represent the formal position of MHLW, PMDA, FDA and USP.
1. USP's Contents of General Chapters
USP -NF General Chapters for Drugs consists of required Chapters for "General Tests and Assays" which are numbered below <1000>, and of informational Chapters as "General Information" which are numbered <1XXX>.
General Chapters for dietary supplements are numbered <2XXX>.
Second Supplement to the USP33-NF28 Reissue has the following contents;
| - General Tests and Assays | |||
| o General Requirements for Tests and Assays | <1> - <11> | ||
| o Apparatus for Tests and Assays | <16> - <41> | ||
| o Microbiological Tests | <51> - <71> | ||
| o Biological Tests and Assays | <81> - <171> | ||
| o Chemical Tests and Assays | <181> - <591> | ||
| ・ Identification Tests | <181> - <201> | ||
| ・ Limit Tests | <206> - <291> | ||
| ・ Other Tests and Assays | <301> - <591> | ||
| o Physical Tests and Determinations | <601> - <941> | ||
| - General Information | <1005> - <1265> | ||
Table of chapter titles (with tentative Japanese translation) is attached.
These Chapters are classified into the following categories:
| 0) | Basic Elements |
| 1) | Non-Complex active Drug Substances |
| 2) | Biotechnology-Derived Drug Substances |
| 3) | Excipients |
| 4) | Non-Complex active Drug Products |
| 5) | Biotechnology-Derived Drug Products |
| 6) | Vaccines |
| 7) | Blood and Blood Products |
| 8) | Gene and Cell Therapy Products |
| 9) | Drug Product Distribution |
| 10) | Microbiology |
| 11) | Dietary Supplement Ingredients |
| 12) | Dietary Supplement Products |
| 13) | Compounding - Substance/Preparation/Practice |
| 14) | Medical Devices |
USP-NF includes the chart of each category which indicates the relation of chapters under the same category.
Example of Chart as Guide to General Chapters
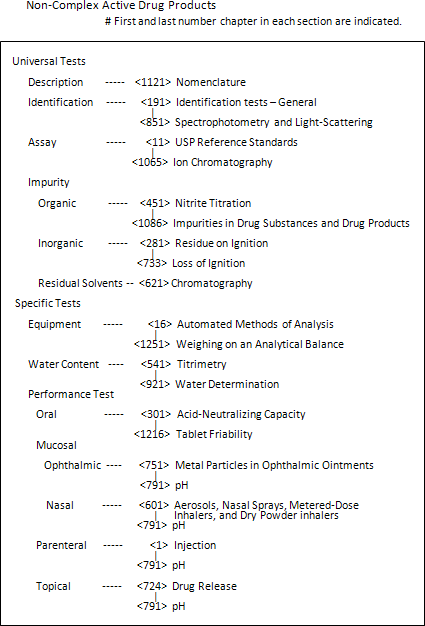
USP-NF introduced the number of each Chapter in 1980 edition (USP20-NF15). Before of that edition, the chapters were in alphabetical order within the each section.
Numbering system of Chapters is somehow similar to numbering of mailing address along the street or address number for computer programming. Most number of main Chapters have <XXX1> and sometimes <XXX5> or <XXX6> to maintain spaces for future chapters. Series of Chapters which were initially established at the same time have consecutive numbers. The number of the deleted Chapter is not used as other Chapter.
The draft chapter number is proposed by the USP secretariat and finalized by the Expert Committee.
2. Standards and Chapters for Compounding
The Compounding Expert Committee oversees USP's standards for compounding pharmacists. These standards help ensure the quality of compounded preparations and the compounding environment, which can lead to safe medication use.
These standards include more than 150 currently official or proposed monographs for compounded preparations, and the following standards in USP General Chapters:
- <795> Pharmaceutical Compounding - Non-sterile Preparations
- <797> Pharmaceutical Compounding - Sterile Preparations
- <823> Radiopharmaceuticals for Positron Emission Tomography Compounding
- <1160> Pharmaceutical Calculations in Prescription Compounding
- <1163> Quality Assurance in Pharmaceutical Compounding
- <1176> Prescription Balance and Volumetric Apparatus
- <1265> Written Prescription Drug Information - Guidelines
All of these chapters can be found in USP-NF and the Pharmacists' Pharmacopeia.
In 2008, USP revised the General Chapter <797> Pharmaceutical Compounding - Sterile Preparations, which was originally published in 2004 to set practice standards to help ensure that compounded sterile preparations are of high quality. This revision was undertaken to strengthen the Chapter, add clarity, and define procedures necessary for compounding sterile preparations focusing on patient safety. This <797> Chapter applies to hospital pharmacies, health care institutions, community pharmacies, etc., where sterile preparations are compounded, stored, and dispensed.
The official version of USP General Chapter <797> Pharmaceutical Compounding - Sterile Preparations is available in the USP-NF, Pharmacists' Pharmacopeia, and the USP <797> Guidebook to Pharmaceutical Compounding - Sterile preparations. The USP <797> Guidebook is an easy-to-read versions of the revised General Chapter <797>, capturing: the Revised General Chapter <797> official on June 1, 2008, General Notices, the USP Revision Process, Enforceability and Recognition of General Chapter <797>, USP Sterile Compounding Expert Committee's response to the comments received on the proposed revision published in 2006.
3. USP Pharmacists' Pharmacopeia
In March 2008 USP published the Second Edition of the USP Pharmacists' Pharmacopeia. This publication combines USP-NF monographs and general chapters along with authorized reference information developed by USP's Council of Experts and is a dedicated reference for pharmacy, and other healthcare practitioners and students. Compiled under the guidance of leading USP experts, this reference provides comprehensive and critical information that will enhance the knowledge of today's practicing pharmacists and compounding professionals.
The latest edition contains:
-129 official USP-NF monographs for compounded preparations,
-73 USP General Chapters covering compounding, packaging, labeling, and storage of pharmaceutical preparations,
-General Chapter <797> Pharmaceutical Compounding-Sterile Preparations,
-Veterinary Compounding and Information,
-Food Ingredients and Flavorings, with more than 500 monographs for preservatives, flavorings, and colorings from the Food Chemicals Codex, Sixth Edition
-Legal requirements and laws applicable to compounding.
The USP Pharmacists' Pharmacopeia can assist pharmacies and pharmacists in both communities and hospitals, setting through
-Serving as a valuable, comprehensive reference on pharmacy and healthcare practices - particularly compounding, packaging, and storage,
-Facilitating compliance with various regulatory standards and procedures required by federal and state laws,
-Introducing best practices in safe and effective preparing and dispensing compounded preparations,
-Serving as a comprehensive, go-to reference on multiple aspects of compounding practice,
-Helping maintain preparedness for regulatory survey and inspection.
4. Medicare Model Guidelines
In December 2003, the Medicare Prescription Drug Improvement and Modernization Act of 2003 (MMA) was signed into law, and the United States Pharmacopeial Convention (USP) is named in Section 1860D-4(b)(3)(C)(ii) of the Act.USP was charged with developing a list of categories and classes that may be used by prescription drug plans and to revise such classification from time to reflect changes in therapeutic uses of covered part D drugs and the additions of new covered part D drugs.
Based on the contract with the Centers for Medicare and Medicaid Services (CMS), in 2004, USP organized the Model Guidelines Expert Committee with 17 scientific experts in various therapeutic fields, established the USP Model Guideline (first version) which included the list of the "USP Category" and the "USP Class" of prescription drugs, and reported to CMS. The CMS utilized such categories and classes for their formulary review process of Medicare Part D Plan.
After 2005, the USP Model Guidelines have been updated several times. In 2008, Centers for Medicare and Medicaid Services (CMS) requested that the USP Model Guidelines be updated on an every three year basis. For 2010-2015 USP cycle, the Model Guidelines Expert Panel was appointed and is responsible for reviewing and updating the Medicare Model Guidelines version 5.0. The experts appointed to this Expert Panel include pharmacists, pharmacologists, health care practitioners, drug information experts and others.
USP Model Guidelines (Example)
| USP Category | USP Class |
|---|---|
| Analgesics | Nonsteroidal Anti-inflammatory Drugs |
| Analgesics | Opioid Analgesics, Long-acting |
| Analgesics | Opioid Analgesics, Short-acting |
| Anesthetics | Local Anesthetics |
| Antibacterials | Tetracyclines |
| Antibacterials | Quinolones |
| Antibacterials | Macrolides |
| Antibacterials | Aminoglycosides |
| Antibacterials | Sulfonamides |
| Antibacterials | Beta-lactam, Penicillins |
| Antibacterials | Beta-lactam, Cephalosporins |
| Antibacterials | Beta-lactam, Other |
| Antibacterials | Antibacterials, Other |
5. State of the Union address by the President
In advance of the President's budget request, President Barack Obama delivered the State of Union address on January 25, 2011. Its formal basis is from the U.S. Constitution, which states that the President shall give to the Congress information on the state of union. This year, the President mostly spoke on his vision and agenda for the American economy and economic revival, and he emphasized the importance of investments in innovation, education, and infrastructure, including biomedical research, information technology, and clean energy technology.
On the other hand, the President addressed reducing health care cost, the freeze of annual domestic spending for the next five years, and a proposal to merge, consolidate, and reorganize the federal government.
On February 14, 2011, the President sent to Congress his FY2012 budget plan, but the Congress is debating the remainder of FY2011 budget request after the current Continuing Resolution. (As of last February)
6. Prescription Drug User Fee
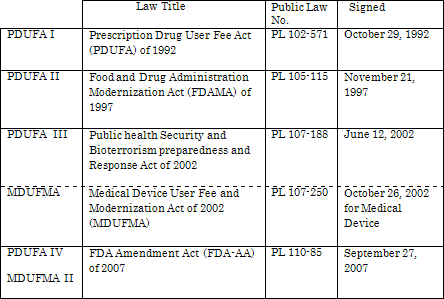
User Fee for the application of the Prescription Drug was introduced in 1992 (at the 1993 Fiscal Year) by the Prescription Drug User Fee Act (PDUFA).
Since the PDUFA is a legislation of five-year duration, it has been renewed in 1997 (PDUFA II), in 2002 (PDUFA III), in 2007 (PDUFA IV), and it will be renewed in 2012.
PDUFA I FY1993 - FY1997 (Oct 1, 1992 - Sep 30, 1997)
PDUFA II FY1998 - FY2002 (Oct 1, 1997 - Sep 30, 2002)
PDUFA III FY2003 - FY2007 (Oct 1, 2002 - Sep 30, 2007)
PDUFA IV FY2008 - FY2012 (Oct 1, 2007 - Sep 30, 2012)
PDUFA V FY2013 - FY2017 (Oct 1, 2012 - Sep 30, 2017) (estimated)
PDUFA I (Public Law 102-571) amended the Federal Food, Drug, and Cosmetic Act (FFDCA), and authorized FDA to collect application fees of new human drugs from the sponsor; pharmaceutical companies.
Basic goal of PDUFA was to shorten the time from the submission of an application to FDA's approval decision, and the performance goals of PDUFA included that FDA would review 55-90%(step by step in each year) of standard applications within 12 months and 90% of Priority applications within 6 months. And it was also to eliminate backlogs of NDA/BLA etc., at that time.
PDUFA established three kinds of prescription drug user fees (one-third of the total fees revenue for each);
-Application review fees : a fee for the review of each new or supplement drug approval application or biologics license application
-Establishment fees : an annual fee for each manufacturing establishments
-Product fees : an annual fee for each products that the PDUFA defines
In 1997, US Congress established the Food and Drug Administration Modernization Act (FDAMA), and it reauthorized the PDUFA for next 5 year cycle.
PDUFA II indicated that the fees must be used for the expenditure of the development of new drug and the review process of new drug application. PDUFA II required the Secretary of Health and Human Services (HHS) to make the goals published in the Congressional Record, and to send annual performance and financial reports to Congress.
In the reauthorization of PDUFA in 1997, the Congress requested performance goals including investigational phase of new drug development (e.g. review of special protocol assessments), transparent drug review process (e.g. response to appeal of decision), and better communication with industries and patient advocacy groups(e.g. response to meeting requests).
The Public Health Security and Bioterrorism Preparedness and Response Act of 2002 included the reauthorization of PDUFA from 2002, and the FDA can adjust annual revenue targets based on their workload changes.
The PDUFA III also allowed FDA to appropriate the fees to support their post-marketing surveillance activities and to develop databases for drug use. It also asked sponsors to include risk management plans in the documents at the pre-NDA/BLA meeting.
Therefore, FDA review activities with user fees cover stages of a drug preclinical development, a clinical development, an application for marketing authorization, and post-marketing safety surveillance and risk management. And the Good Review Management Principles and Practices (GRMPs) developed under PDUFA III described an opportunity of FDA-sponsor interactions during first review cycle.
In 2007, the Congress passed the FDA Amendment Act (FDA-AA), and reauthorized and expanded the PDUFA.
PDUFA IV (from FY2008 to FY2012) emphasizes drug safety activities and recommends the following three items;
-Ensuring sound financial footing,
-Enhancing the process for pre-market review,
-Modernizing and Transforming the Post-market Drug Safety System.
FDA established and updated the Drug Safety Five Year Plan, which includes assessment of methodologies to maximize the public health benefit associated with collecting adverse events, identification of epidemiology best practices and developing guidance, expanding database acquisition and use for the purposes of targeted post-marketing surveillance and epidemiology, developing and validating risk management and risk communication tools, improving post-market IT systems, enhancing and improving communication and coordination between the offices in CDER and CBER, review of proprietary names to reduce medication errors.
FDA-AA also reauthorized the Best Pharmaceuticals for Children Act (BPCA) enacted in 2002, and the Pediatric Research Equity Act (PREA) enacted in 2003, both of which are designed to encourage more research into, and more development of, treatments for children.
7. Prescription Drug User Fee Rate and Revenue
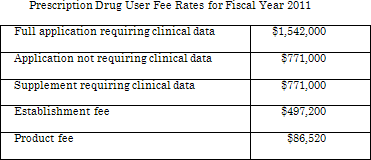
Prescription Drug User Fee Rates for FY 2011 was announced by the Federal Register issued on August 4, 2010. The User Fee Rates are based on the total fee revenue amount for FY 2011 with base adjustment to statutory fee revenue amount, inflation adjustment, workload adjustment, and rent and rent-related adjustment. The application fee, the establishment fee, and product fee is set to respectively generate one-third of the total fee revenue amount in FY 2011($206,356,667). The fee-paying full application equivalents (FAEs) are estimated as five-year average, and the application fee is calculated by dividing the FAEs(133.8) into the fee revenue amount to be derived from application fees.
The result, rounded to the nearest $100, is a fee of
-$1,542,000 per full application requiring clinical data,
-$771,000 per application not requiring clinical data,
-$771,000 per supplement requiring clinical data.
The number of establishments that would be subject to, and would pay fees, is estimated (415). The fee per establishment is determined by dividing the fee revenue amount to be derived from establishments ($206,356,667) by the estimated 415 establishments, for an establishment fee rate of $497,200 (rounded to the nearest $100).
The number of product that would be subject to, and would pay product fees, is 2,385. The FY 2011 product fee rate is determined by dividing the fee revenue amount to be derived from product fees ($206,356,667) by the estimated 2,385 products for a product fee of $86,520 (rounded to the nearest $10).
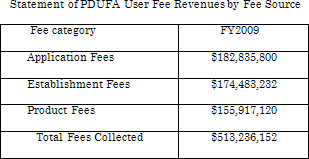
According to the FY2009 PDUFA Financial Report, total fees collected in FY2009 (as of September 30, 2009) is $513,236,152. The following table provides a breakout of user fees collected by fee category.
User fee revenues are expended only for costs necessary to support the process for the review of human drug applications, as defined in PDUFA. In FY 2009, FDA obligated $512,051,400 from prescription drug user fee revenues.
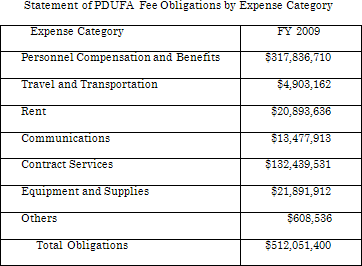
Of the total of $855,426,294 obligated in support of the process for the review of human drug applications in CDER, CBER, ORA, OC as defined in PDUFA in FY 2009, about 60 percent came from PDUFA fees and about 40 percent came from Federal Government appropriations.

Now, 65 percent or more of human drug review funding comes from user fees. Since the legislative authority for PDUFA expires in September 2012, FDA held the public meeting on the PDUFA on April 12, 2010, to share assessment of the overall performance of the PDUFA IV program thus far, and to hear what aspects of PDUFA should be retained, changed, or discontinued.
8. Medical Device User Fee
The Medical Device User Fee and Modernization Act of 2002 (MDUFMA), (P.L 107-250), amends the Federal Food, Drug, and Cosmetic Act (FD&C Act) to provide FDA new responsibilities, resources, and challenges. MDUFMA was signed into law October 26, 2002. The MDUFMA was amended by the Medical Devices Technical Corrections Act (MDTCA), P.L 108-214 in 2004.
MDUFMA has four particularly significant features:
- User fees for premarket reviews of PMAs, PDPs, premarket reports (a new category of premarket application for reprocessed single-use devices), BLAs, certain supplements, and 510(k)s.
- Performance goals for many types of premarket reviews. These goals become more demanding over time, and include FDA decision goals and cycle goals (cycle goals refer to FDA actions prior to their final action on a submission).
- Establishment inspections may be conducted by accredited persons (third-parties), under carefully prescribed conditions.
- New regulatory requirements for reprocessed single-use devices, including a new category of premarket submission, the premarket report.
The Medical Device User Fee and Modernization Act (MDUFMA) have been reauthorized and expanded by the Food and Drug Administration Amendments Act of 2007. MDUFA II established the specific timelines for Modular PMAs and Real-Time PMA supplements.
9. Medical Device User Fee Rate and Revenue
The act (MDUFMA) specifies the standard fee for a premarket approval application (PMA) for each year: the standard fee for a premarket application received by FDA during FY 2011 is $236,298. The fee rate for each type of submission is also set by the act at a specified percentage of the standard fee for a premarket application as follows.
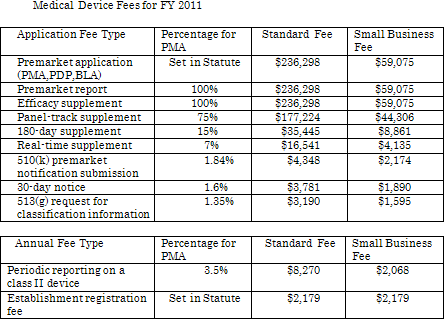
If the applicant has gross receipts or sales of no more than $100 million for the most-recent tax year, it may qualify for reduced small business fees. If the applicant has gross receipts or sales of no more than $30 million, it may qualify for a waiver of the fee for its first premarket application (PMA, PDP, BLA) or premarket report.
The act also sets the annual fee for establishment registration at $2,179 in FY 2011, and there is no reduced fee rate of annual establishment registration for small business.
The net revenues of user fees in FY 2009 by category of source are as follows.
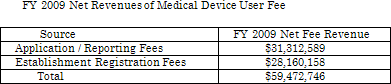
The legislative authority for the medical device user fee program expires in September 2012. At that time, new legislation will be required for FDA to continue collecting user fees for the medical device program. FDA held the public workshop for Medical Device User Fee Program on September 14, 2010, to hear stakeholder view on medical device user fee reauthorization.
10. Drug Safety Five-Year Plan under the Prescription Drug User Fee Act (PDUFA) IV
FDA completed the Prescription Drug User Fee Act (PDUFA) IV Drug Safety Five-Year Plan in December 2008. The plan detailed strategies for improving FDA's expertise in monitoring drug/biologics product safety.
FDA established post-marketing drug safety performance goals, described in the PDUFA Reauthorization Performance Goals and Procedures (FY2008-FY2012) in Section VIII (Enhancement and Modernization of the FDA Drug Safety System) and Section IX (Review of Proprietary Names to Reduce Medication Errors). The plan includes each commitment, its background and recent accomplishment.
Followings are the main accomplishments described in the 2009 Update of Activities of the plan and their follow up.
| - | "The plan - 2009 update" is the first annual assessment, describing progress on PDUFA IV drug safety commitments through the end of FY 2009. | ||
| - | After receiving and reviewing the requests for information (RFIs), the request for proposals for research on how to maximize the benefit of collecting spontaneous adverse event reports information was issued on July 17, 2009. The evaluation of the value and contribution of FDA's adverse event surveillance system to safety-related regulatory actions is underway. | ||
| - | A public workshop, "Developing Guidance on Conducting Scientifically Sound Pharmacoepidemiologic Safety Studies using Large Electronic healthcare Data Sets," was held on May 7, 2008, and the CDER/CBER Work Group reviewed input from the workshop and subsequent comments in order to prepare the final draft guidance. | ||
| - | In order to expand CDER/CBER's database acquisition and use for the purposes of targeted post-marketing surveillance and epidemiology, | ||
| 1) | CDER Program with Centers for Medicare and Medicaid Services (CMS) | ||
| i) | Fluoroquinolones and Acute Achilles Tendon Rupture Risk Study | ||
| ii) | Fracture risk, all-cause mortality, and cardiac arrhythmias with the use of propoxyphene and other analgesics | ||
| iii) | Potential interactions between concurrent use of two anti-diabetic drugs (rosiglitazone and pioglitazone) and fibrates (cholesterol-reducing drugs) and the risk of heart attack, stroke, and death in elderly patients with type 2 diabetes. | ||
| iv) | A study of exenatide compared to sitagliptin (different types of drugs that are both indicated for the treatment of type 2 diabetes) and the risk of severe acute pancreatitis in the elderly. | ||
| 2) | CDER Program with Agency for Healthcare Research and Quality (AHRQ) | ||
| i) | A comparative assessment of the safety of antipsychotic agents. | ||
| ii) | Tissue necrosis factor (TNF) blockers (agents used to treat inflammatory conditions) and multiple safety outcomes. | ||
| iii) | Patterns of acetaminophen use and knowledge of safety issues. | ||
| iv) | Pediatric inpatient drug use data and feasibility of making national projections. | ||
| 3) | CDER Program with Veterans Health Administration (VHA) | ||
| i) | A study of HMG-CoA reductase inhibitor (statins) use and risk of amyotrophic lateral sclerosis (ALS) has received institutional review board (IRB) approval and is now launched. | ||
| ii) | Antipsychotic use and risk of mortality. | ||
| iii) | Antipsychotic use and differential risks of tardive dyskinesia (abnormal movements). | ||
| iv) | Varenicline (a drug used as a smoking cessation aid) and the risk of serious psychiatric disorders (done in partnership with FDA, VHA and DoD). VHA IRB approval for this study has been obtained. DoD IRB review is underway, as are DoD approval processes for medical record verification. | ||
| 4) | CDER Program with Department of Defense (DoD) | ||
| i) | Software enhancement for drug safety signal identification and confirmation via epidemiologic studies. | ||
| 5) | CDER Program with Centers for Disease Control and Prevention (CDC) | ||
| i) | Under an interagency agreement, FDA provided funds to CDC to develop a user-friendly query tool for National Electronic Injury Surveillance System Cooperative Adverse Drug Event Surveillance (NIESS-CADES), an emergency medicine database, for more efficient use by CDER's Office of Surveillance and Epidemiology (OSE) safety evaluators and epidemiologists. | ||
| ii) | Selected review and data collection projects using NEISS or NEISS-CADES in 2009 include intravenous iron and allergic reactions, accidental ingestion of opioids by children, and acetaminophen-related emergency department visits. | ||
| 6) | CDER Program with Private Collaborators (Ingenix, Inc., Kaise Foundation Research institute, Vanderbilt University, HMO Research Network) | ||
| i) | A multi-site study of drugs used to treat attention deficit/hyperactivity disorder (ADHD) and possible associated risks for serious cardiovascular outcomes (sudden cardiac death, heart attack, or stroke) in children. | ||
| ii) | Two studies on oral bisphosphonate use : (1) bisphosphonates and associated risk of atrial fibrillation (AF), and (2) investigation into the prevalence of and risk factors for osteonecrosis of the jaw among users of bisphosphonates. | ||
| iii) | Data collection completed for a multi-site study of newer combined hormonal contraceptive products to examine their risk for blood clot-related events such as sudden death, heat attack, pulmonary embolism and deep vein thrombosis. | ||
| iv) | Three feasibility studies in a multi-site program entitled the Medication Exposure in Pregnancy Risk Evaluation program (MEPREP) were underway in FY 2009 to initiate data linkages for studies of drugs used during pregnancy and potential adverse outcomes in the newborn. | ||
| 7) | CBER Initiatives | ||
| i) | The Flu Vaccine Safety Project with CMS to use Medicare data for active surveillance of influenza vaccine safety. CBER developed methods to perform rapid rate-based monitoring of adverse events among millions of vaccines. |
||
| ii) | CBER/CDC Vaccine Safety Data-link Inter-Agency Agreement for near-real time surveillance of vaccine adverse events. The Vaccine Safety Data-link (VSD) monitors immunization safety and serves as an informational and research resource for rare and serious post-immunization events. FDA joins the VSD's existing collaboration between CDC's Immunization Safety Office and eight managed care organizations (MCOs). FDA staff collaborated with CDC and VSD investigators in decisions regarding outcomes for pandemic H1N1 vaccine during the 2009-2010 influenza season. CBER staff worked with CDC in determining priorities for specific Rapid Cycle Analysis (RCA) projects, including evaluation of the safety of the pandemic H1N1 influenza vaccine, the HPV vaccine, the measles-mumps-rubella-varicella (MMRV) vaccine, pneumococcal conjugate vaccine (PCV13), and varicella zoster vaccine. |
||
| - | The Risk Communication Advisory Committee reviewed and evaluated public communication programs and strategies concerning risks and benefits of FDA-regulated products, and advised the agency on matters related to public awareness of drug safety issues. | ||
| - | A public meeting on the development and implementation of risk evaluation and mitigation strategies (REMS) for drugs and biological products was held on July 27-28, 2010. FDA obtained input from a wide range of stakeholders about the implementation of the REMS program and the development of guidance documents. | ||
| - | The first prototype of FDA Adverse Event Reporting System (FAERS) has been delivered. The goal of FAERS is to improve quality, usability and efficiency of the passive (voluntary) reporting of a drug's adverse events (AEs). This system in CDER, CBER, CDRH will facilitate the process of bringing events to the appropriate analysts' attention sooner. | ||
| - | Document Archiving Reporting and Regulatory Tracking System (DARRTS) included additional categories to enhance tracking of public notification of safety issues. FDA staffs regularly examine the AERS database as part of routine safety monitoring. When a potential signal of a serious risk is identified from AERS data, it is entered as a safety issue into CDER's Document Archiving, Reporting, and Regulatory Tracking System (DARRTS) or into CBER's Therapeutics and Blood Safety Branch Safety Signal Tracking (SST) system. | ||
| - | CDER and CBER researchers in epidemiology and surveillance studies used the General Practice Research Database (GPRD), including : | ||
| i) | Maternal exposure to antidepressant drugs and Persistent Pulmonary Hypertension in the Newborn (PPHN) | ||
| ii) | Anti-diabetic agents and the risk of cancer | ||
| iii) | Novel approached for validation of PPHN cases in the GPRD | ||
| - | FDA developed a computerized method to determine orthographic (spelling) or phonetic (sound) similarities between proposed proprietary drug names that might increase risks of confusion, medication errors, and inappropriate dosing of medicine. The Phonetic and Orthographic Computer Analysis (POCA) system is an automated method of identifying potential look-alike and sound-alike proprietary and established names. The notice of availability of the source code and supporting technical documentation was published in the Federal Register in 2009. | ||
| - | The final guidance, Contents of a Complete Submission for the Evaluation of Proprietary Names, was published in February 2010. | ||
| - | The CBER Standard Operating Procedures and Policies (SOPP) and the CDER Manual of Policy and Procedure (MAPP) were published in 2008 and 2009 respectively, including internal procedures for submission of proprietary names submitted with INDs, and NDAs/BLAs. | ||
| - | A public workshop, Developing Guidance on Naming, Labeling, and packaging Practices to Minimize Medication Errors, was held in June 2010. The dialogue among regulators, researchers, pharmaceutical industries, health care organizations, health care professionals, and others from the general public is used to develop draft guidance. | ||