
2011年2月28日
植村 展生
2010年2月中旬から、米国東海岸のワシントンDC近郊(正確には、ワシントンDCから地下鉄のメトロでいけるメリーランド州ロックビル市)にある、USP(United States Pharmacopeia:米国薬局方)事務局(以下「USP」という)に、MHLW/PMDAのLiaison Officialとして派遣され、USPのVisiting Scientistとして米国薬局方の総則のチーム内に籍を置いております。
12月27日付けの前号に引き続き、USPとFDAの活動の概要をお伝えします。
なお、この便りは、USPに派遣されている植村がUSP、FDA等に関する情報を個人の立場でとりまとめたものであり、USP、FDA等の米国関係機関あるいは派遣元である厚生労働省(MHLW)、医薬品医療機器総合機構(PMDA)の見解等を示すものではないことにご留意下さい。
また、日本語版は英文版よりも多少言葉を補っていますので、同一の翻訳ではないことをご承知おきください。
第6回ワシントンDCメトロ便り
目次
- 総則(総則と一般試験法)の構成
- 調剤(薬局製剤)の基準
- USP薬剤師薬局方
- メディケアモデルガイドライン
- 大統領一般教書演説
- 処方せん薬ユーザーフィー
- 処方せん薬ユーザーフィーの手数料と収入
- 医療機器ユーザーフィー
- 医療機器ユーザーフィーの手数料と収入
- 第4次処方せん薬ユーザーフィー法に基づく医薬品安全対策5カ年計画
1.総則(総則と一般試験法)の構成
米国薬局方-国民医薬品集(USP-NF)の医薬品総則(総則と一般試験法)は章の番号が1000番以下の法的要件となる試験法・定量法の章と、章番号が1000番台の一般的情報を掲載した参考情報の章とから構成されています。また、総則の2000番台は栄養補助食品の章となっています。
USP33-NF28(再発行)の第二追補版では、総則の構成は以下のようになっています。
| - 一般試験法・定量法 | |
|
<1> - <11> |
|
<16> - <41> |
|
<51> - <71> |
|
<81> - <171> |
|
<181> - <591> |
|
<181> - <201> |
|
<206> - <291> |
|
<301> - <591> |
|
<601> - <941> |
| - 参考情報項目 | <1005> - <1265> |
総則の各項目の一覧表(原本の英語と日本語仮訳)は別添を参照ください。
これらの総則の各章は、以下の項目に分類され体系付けられています。
0) 基本的要件
1) 医薬品有効成分(複合していないもの)
2) バイオテクノロジー由来の医薬品成分
3) 医薬品添加物
4) 有効成分を含む医薬品製品(複合していないもの)
5) バイオテクノロジー由来の医薬品製品
6) ワクチン
7) 血液及び血液製剤
8) 遺伝子、細胞治療製品
9) 医薬品製品流通
10)微生物学
11)栄養補助食品の成分
12)栄養補助食品の製品
13)調剤-成分、製剤、実務
14)医療機器(医療材料)
USP-NFでは分類項目ごとに同じ分類に属する章の関係を図示した体系図を総則のガイドとして掲載しています。
その一例は以下のようなものです。
総則ガイドの例

USP-NFが各総則の章に番号をつけるようになったのは1980年のUSP20-NF15の版からです。それまでの版では、総則の章は各セクションの中で、単純にアルファベット順に並べられていました。
総則の番号付けのシステムは郵便のあて先の通りの住所番号やコンピュータープログラミングの番地の番号と似ています。すなわち、多くの主たる章は末尾が1(<XXX1>)の番号が付されており、その後に追加された章は、将来の更なる追加のためのスペースを残しておくために末尾が5又は6の番号が付されているものが多くなっています。同時期に始めて設定された一連の章については、連番の番号が付されています。削除された章の番号は他の章に使用されることはありません。この総則の章の番号は、新たに設定する際には、USPの事務局が原案を作成し、専門家委員会で最終的に決定されます。
2.調剤(薬局製剤)の基準
USPの調剤専門家委員会は調剤業務を行う薬剤師のためにUSPが設定する基準を所管しています。この基準は、調剤された製剤の品質や調剤業務を行う環境の水準を確保し、そのことによって薬剤の安全な使用に寄与するものです。
このような調剤関係の基準としては、調剤された製剤の医薬品各条が提案中のものも含めて現在約150品目以上示されている他、総則の中に以下の章が設定されています。
<795> 調剤(薬局製剤)-非無菌製剤
<797> 調剤(薬局製剤)-無菌製剤
<823> PET用放射性医薬品
<1160> 処方せん調剤における薬の計算
<1163> 医薬品調剤における品質保証
<1176> 調剤用はかり、調剤用計量器
<1265> 処方せん薬文書情報-ガイドライン
これらの総則各章は、公定書である米国薬局方-国民医薬品集(USP-NF)と薬剤師薬局方に掲載されています。
2008年にUSPは総則第797章「医薬品調剤-無菌製剤」の改訂を行いました。この章は、無菌製剤の調製を高い品質レベルで行うことができるように、その実施基準を定めるため2004年に設定されたものです。2008年の改訂では、章の内容を強化し、明確にし、そして、患者の安全性に重点を置いた無菌製剤の調製のために必要な手順を規定することが行われました。この第797章は、病院内の薬局、医療施設、調剤薬局など、無菌製剤の調製が行われ、その製剤が保存され、患者に渡される施設に適用されるものです。
公式のUSP総則第797章「医薬品調剤-無菌製剤」は米国薬局方―国民医薬品集(USP-NF)に掲載されていますが、薬剤師薬局方、USP第797章「医薬品調剤-無菌製剤」ガイドブックにも掲載されています。このUSP第797章ガイドブックは読者に分かりやすく編集されたもので、改訂され2008年6月1日に発効した第797章を含む他、通則、USPの改訂手順、総則797章の施行やそれによる許可の範囲、2006年に公表された改定提案に対して寄せられた意見に対するUSP無菌調剤専門家委員会の回答が掲載されています。
3.USP薬剤師薬局方
2008年3月、USPは第二版のUSP薬剤師薬局方を出版しました。これは、USPの評議会の定めたところによる関係情報に沿って米国薬局方-国民医薬品集(USP-NF)の関係各条と総則を統合したものであり、薬局その他の医療関係者や学生にとって手引きとなるものです。USPの専門家によるガイダンスに沿って編纂されたこの手引書は、今日の薬剤師業務や専門的調剤業務を行う者の知識を高める包括的かつ重要な情報を提供するものです。
最新版の薬剤師薬局方には、以下の内容が含まれています。
- 調剤された製剤についての129品目のUSP-NF各条
- 医薬品製剤についての調剤、包装、表示、保管に関する73の章の総則
- 総則第797章 調剤(薬局製剤)-無菌製剤
- 動物用医薬品の調剤と関連情報
- 食品内成分と香料;Food Chemical CODEX第6版に基づく500品目以上の保存料、香料、色素
- 調剤に関する法的要件と法の適用
USP薬剤師薬局方は、以下のことを通して、薬局そして地域の薬局と病院内の薬局で働く薬剤師を支援するものです。
- 薬局、医療施設での実務、特に調剤、包装、保管に関して重要かつ包括的な情報を含んだ手引きとして利用される。
- 連邦法及び州法によって要求される様々な規制基準や手順の遵守を促進する。
- 調剤される製剤について、安全性及び有効性の確保された調剤準備、調剤と患者への分配の際の適切な手順の導入を図る。
- 調剤業務の様々な場面での総括的かつ牽引的な手引きとして利用される。
- 規制当局による調査や査察に対する準備のレベルを保持する助けとなる。
4.メディケアモデルガイドライン
2003年12月、2003年メディケア処方せん薬改善近代化法(MMA)が署名されて法律として発効しましたが、その中(セクション 1860D-4(b)(3)(C)(ii))に、米国薬局方Convention(USP)の名前が記述されました。すなわち、メディケアの処方せん薬計画において使用される処方せん薬の分類と区分のリストを作成することと、パートD医薬品の臨床使用の変更や新たにカバーされるパートD医薬品の追加に対応して、そのリストの分類を修正することが義務付けられました。
メディケア・メディケイドサービスセンター(CMS)との契約に基づき、USPは2004年に、17人の様々な医療分野の専門家からなるモデルガイドライン専門家委員会を組織し、USPによる処方せん薬の分類と区分のリストを含めた第一版のUSPモデルガイドラインを作成して、CMSに報告しました。CMSはこのUSPの分類と区分をメディケアパートD計画の採用医薬品集の見直しプロセスに利用しています。
2005年以降、USPモデルガイドラインは数回改訂されました。2008年には、メディケア・メディケイドサービスセンター(CMS)から、3年毎の頻度での改訂が要求され、USPでは2010-2015年サイクルでの活動において、モデルガイドライン専門家パネルを設置し、第5.0版のモデルガイドラインへの改定作業を受け持っています。この専門家パネルには、薬剤師、薬理学者、医療関係者、薬剤情報専門家などの専門家が指名され参加しています。
USPモデルガイドラインの例(一部抜粋)
| USP 分類 | USP 区分 |
|---|---|
| 鎮痛薬 | 非ステロイド性抗炎症薬 |
| 鎮痛薬 | 麻薬性鎮痛薬、長時間作用型 |
| 鎮痛薬 | 麻薬性鎮痛薬、短時間作用型 |
| 麻酔薬 | 局所麻酔薬 |
| 抗菌性製剤 | テトラサイクリン系 |
| 抗菌性製剤 | キノロン系 |
| 抗菌性製剤 | マクロライド系 |
| 抗菌性製剤 | アミノグリコシド系 |
| 抗菌性製剤 | スルフォンアミド系 |
| 抗菌性製剤 | ベータラクタム、ペニシリン系 |
| 抗菌性製剤 | ベータラクタム、セファロスポリン系 |
| 抗菌性製剤 | ベータラクタム、その他 |
| 抗菌性製剤 | 抗菌性製剤、その他 |
5.大統領一般教書演説
2012会計年度大統領予算教書の発表に先立ち、オバマ大統領は1月25日に米国議会で一般教書演説を行いました。この一般教書演説の根拠は、米国憲法に述べられており、大統領は議会に対して国の現状に関する情報を示すことができるとされていることによるものです。今年の一般教書演説では、主に米国経済の競争力低下対策が語られる中で、イノベーション、教育、インフラストラクチャへの投資、その中では、生命科学研究や情報技術、クリーンエネルギーへの投資等の重要性が指摘されましたが、その一方で、大統領は医療コストの削減、歳出の5年間の据え置きと連邦政府の組織改革の提案にも言及しています。
大統領は2012会計年度の予算教書を2月14日に議会に提出しましたが、議会では2011会計年度の継続予算後の残りの予算の議論が続いています。(2月末現在)
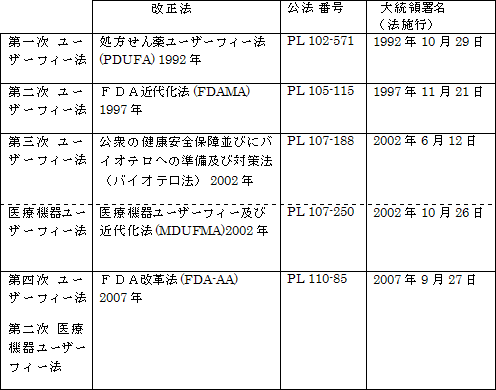
6.処方せん薬ユーザーフィー
処方せん薬の申請に関するユーザーフィー(手数料)は処方せん薬ユーザーフィー法(PDUFA)の成立によって1992年(1993会計年度)から導入されました。
この処方せん薬ユーザーフィー法は5年間の時限立法のため、これまでにその改正が1997年(PDUFA II)、2002年(PDUFA III)、2007年(PDUFA IV)に行われ、おそらく2012年に次の改正が行われることになります。
PDUFA I 1993 - 1997会計年度 (1992年10月1日 - 1997年9月30日)
PDUFA II 1998 - 2002会計年度 (1997年10月1日 - 2002年9月30日)
PDUFA III 2003 - 2007会計年度 (2002年10月1日 - 2007年9月30日)
PDUFA IV 2008 - 2012会計年度 (2007年10月1日 - 2012年9月30日)
PDUFA V 2013 - 2017会計年度 (2012年10月1日 - 2017年9月30日) (推定)
第一次のユーザーフィー法(公法:PL102-571)は連邦食品医薬品化粧品法を改正する法律として成立し、人用の新医薬品の申請手数料を申請者、すなわち製薬企業から徴収する権限をFDAに初めて与えたものです。
ユーザーフィー法の基本的な目的は、新医薬品の申請書の提出からFDAによる承認の判断までの期間を短縮することにあり、この第一次ユーザーフィー法に基づいて設定された業績目標の中には、新薬申請の標準審査の期間が12ヶ月以内であった割合の目標を55%から段階的に90%とすること、優先審査の期間が6ヶ月以内であった割合の目標を段階的に90%とすることが盛り込まれています。また、申請の滞貨を一定期間に処理することも含まれました。
処方せん薬ユーザーフィー法は以下の3種類の申請料の区分を設定し、それぞれの区分が総収入の3分の1をカバーする申請料となっています。
1)申請、追加申請手数料:新薬承認申請又は追加申請(変更承認申請)、生物製品新規許可申請又は追加(変更)許可申請
2)施設手数料:各製造施設ごとの年間手数料
3)製品手数料:法令で定められた各製品ごとの年間手数料
1997年に米国議会はFDA近代化法を成立させ、処方せん薬ユーザーフィー法の次の5年間の期間への適用を認めました。このPDUFA IIでは手数料収入は新薬の開発と新薬申請審査の過程に使用されなければならないことが明記され、米国保健福祉長官に対し、議会への報告書の中に目標を示すことと、業績報告及び財政報告を毎年議会に提出することを求めています。
1997年の再認可では米国議会は、新薬の開発段階での治験における対応(例えば特別プロトコール査定の審査)、審査過程の透明化(例えば、申し立てに対する対応)、企業や患者支援団体との対話の改善(例えば面談の要請に対する対応)などの業績目標を求めました。
2002年のバイオテロ法(公衆の健康安全保障並びにバイオテロへの準備及び対策法)は、その中に2002年からのユーザーフィー法の再認可を含んでおり、また、この中でFDAは年間収入の目標をFDAの作業量に基づいて調整できることが盛り込まれました。また、このPDUFA IIIは、FDAが申請料を市販後監視業務の支援や医薬品の使用に関するデータベースの開発に当てることができるようになりました。さらに、申請者に対しては申請前相談における提出文書の中にリスクマネージメント計画を含めることを求めました。
このような経過を経て、FDAのユーザーフィーを用いた審査業務は医薬品の前臨床試験、臨床試験、承認申請、市販後監視とリスクマネージメントの各段階をカバーすることができるようになりました。また、第3次ユーザーフィー法に基づいて策定された審査の運営方針と作業に関する規範(審査マニュアル)は初回審査過程におけるFDAと申請者の間の対話の機会について明記しています。
2007年には米国議会はFDA改革法(FDAAA)を可決し、その中でユーザーフィー法の再認可と適用拡大を認めています。
PDUFA IV(2008会計年度から2012会計年度まで)は医薬品の安全対策に力点をおいており、以下の3点を勧告しています。
- 財政基盤の健全性確保
- 承認前審査過程の強化
- 医薬品市販後安全性確保システムの近代化と変革
FDAは医薬品安全対策5カ年計画を策定し、その内容を更新していますが、その中で、副作用情報の収集によって公衆衛生の効用の最大化を図ることのできる手法の開発評価、薬剤疫学の効果的手法の確定とガイダンスの開発、データベースの取り扱い範囲の拡大と市販語幹氏や薬剤疫学の目的への利用、リスクマネージメントやリスクコミュニケーションの手法の開発と評価、市販後ITシステムの改善、CDERとCBERの間でのコミュニケーションと連絡調整の改善、薬剤過誤を減らすための商品名(ブランド名)の審査などを示しています。
なお、FDA改革法は、2002年に発効した小児最良医薬品法(BPCA)や、2003年に発効した小児科研究均等法(PREA)の内容を含んであり、小児の治療法についての研究の促進と一層の開発を目指したものとなっています。
7.処方せん薬ユーザーフィーの手数料と収入
2011会計年度における処方せん薬ユーザーフィーの手数料(単価)については、2010年8月4日の官報(FR)で公示されました。ユーザーフィーの手数料単価は、もともと法律に示された補正後の手数料総収入に、インフレーションによる補正、作業量による補正、賃貸料及びその関連による補正を加味した手数料総収入に基づいて算出されます。そして、申請手数料、施設手数料、製品手数料は、それぞれ各項目の収入が2011会計年度の総収入見込み(206,356,667ドル)の3分の1ずつの額を確保するように設定されます。申請件数については、手数料の支払いを伴った新薬の新規(フル)申請に換算した数(FAEs)が、過去5年間の平均から推定されます。そしてこの推定申請件数(2011会計年度は133.8件)を用いて、申請手数料で得られる収入見込額を割り返すことにより、新薬の申請手数料の単価が算出されます。端数を100ドル単位で四捨五入して以下のように算出されました。
臨床試験データの提出を伴う新薬新規(フル)申請 1,542,000ドル
臨床試験データの提出を伴わない新薬申請 771,000ドル
臨床試験データの提出を伴う新薬の追加(変更)申請 771,000ドル
手数料の支払い対象となる施設の数は、2011会計年度では415施設と推定されました。1施設あたりの手数料単価は、施設手数料で得られる収入見込額(206,356,667ドル)を推定施設数で割り返し、100ドル単位で四捨五入して、2011会計年度では497,200ドル)と算出されました。
手数料の支払い対象と指定される製品の数は、2011会計年度では2,385品目と推定されました。1製品あたりの製品単価は、製品手数料で得られる収入見込額(206,356,667ドル)を推定品目数(2,385)で割り返し、10ドル単位で四捨五入して、2011会計年度では、86,520ドルと算出されました。

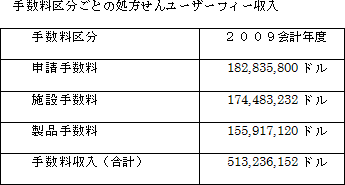
2009会計年度処方せんユーザーフィー法財務報告によると、2009会計年度(2009年9月30日までの時点)に集められた手数料は総額で513,236,152ドルでした。各手数料区分ごとの内訳は以下の表のとおりです。
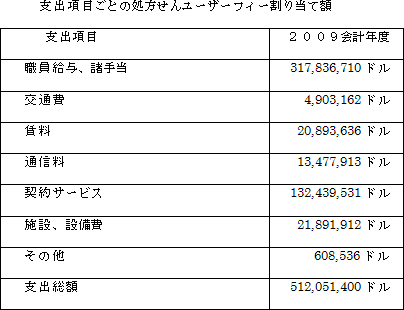
処方せんユーザーフィー法の規定するところに基づき、その手数料収入は人用医薬品の新薬申請の審査過程を支援するのに必要な経費にのみ支出されることとされています。2009会計年度では、FDAは処方せんユーザーフィーの収入から512,051,400ドルを支出にあてました。
処方せんユーザーフィー法の規定するところに基づき、2009会計年度に人用医薬品の新薬申請審査の過程の支援のために医薬品評価研究センター(CDER),生物製品評価研究センター(CBER),規制業務部(ORA),長官事務局(OC)に割り当てられた支出の総額は855,426,294でしたが、そのうちの約60%は処方せん薬ユーサーフィーからのものであり、約40%が連邦政府予算から充当された分でした。

現在(2010年時点)、ユーザーフィーから充当されている人用医薬品新薬申請審査経費の中の割合は65%を超えるレベルとなっています。現在のユーザーフィー法の法的権限は2012年9月で期限を迎えますので、FDAは2010年4月12日に公開の会議を開き、これまでの第四次ユーザーフィー法に基づく活動の実績に対する評価と、次のユーザーフィー法では何を継続し、何を変更し、何を中止すべきかの意見を広く一般から聴取しました。
8.医療機器ユーザーフィー
2002年の医療機器ユーザーフィー・近代化法(医療機器ユーザーフィー法:MDUFMA)(公法PL107-250)によって連邦食品医薬品化粧品法(FD&C法)が改正され、FDAに対して医療機器ユーザーフィーに関する新たな責務と資金と活動を課しました。この医療機器ユーザーフィー法は2002年10月26日に大統領が署名して発効しましたが、その後2004年の医療機器技術修正法(MDTCA)(公法PL108-214)によって修正が行われています。
医療機器ユーザーフィー法は特に重要な以下の4項目の内容を含んでいます。
- 新医療機器申請、製品プロトコール申請、市販前報告(再加工された単回使用製品の市販前申請として新たに設けられた項目)、生物製品許可申請、追加(変更)申請、510(k)届出に対し、それらの市販前審査に関する手数料の設定。
- 各種市販前審査の業績目標の設定。この目標は、審査時間を一層要求し、また、FDAの判断の目標、中間目標(中間目標とは提出された申請に対する最終判断の前のFDAの対応)を含むものとなっています。
- 施設の査察は、厳密な条件の下で、認定された者(第三者機関)が行うことも可能となったこと。
- 再加工された単回使用製品について、市販前申請や市販前報告といった新たな規制要件項目が設けられたこと。
医療機器ユーザーフィー法は、2007年のFDA改革法によって再認可されると同時に適用が拡大され、第二次医療機器ユーザーフィー法では、モジュール申請やリアルタイム変更申請についてのタイムライン(処理時間)が設けられました。
9.医療機器ユーザーフィーの手数料と収入
医療機器ユーザーフィー法は新医療機器承認申請手数料の基準額を法で定めています。2011会計年度にFDAが徴収する新医療機器市販前申請手数料の基準額は236,298ドルとなっています。各区分ごとの手数料(申請手数料、年間手数料)は同法で新医療機器申請手数料に対する比率(パーセンテージ)で以下のように設定されています。
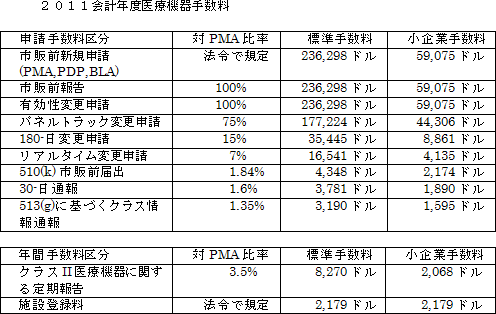
直近の課税年における総収入又は総売り上げが1億ドル又はそれ以下の申請者の場合は、減額された小企業手数料の適用を受けることができます。同じく3000万ドル以下の場合は最初の新医療機器申請(PMA, PDP, BLA)又は市販前報告の申請の手数料の免除を受けることができます。
医療機器ユーザーフィー法は施設登録の年間手数料も規定しており、2011会計年度では2,179ドルが設定されています。施設登録年間手数料には小企業に対する減額された手数料は設けられていません。
2009会計年度における医療機器ユーザーフィーの区分ごとの収入(純収入)は以下のとおりです。

医療機器ユーザーフィー法に基づく活動の法的権限は同じく2012年9月に期限を迎えますので、その時点には、新たな法律によってFDAが医療機器の活動のためのユーザフィーを引き続き徴収できることが求められることになります。FDAは2010年9月14日に公開のワークショップを開催し、医療機器ユーザーフィーの再認可に関する関係者の意見を聴取しました。
10.第4次処方せん薬ユーザーフィー法に基づく医薬品安全対策5カ年計画
2008年12月、FDAは第4次処方せん薬ユーザーフィー法医薬品安全対策5ヵ年計画を策定しました。この計画は医薬品及び生物製品の安全性をモニターするに当たってFDAの専門性をより一層活用するための戦略を詳細にまとめたものです。
FDAは処方せん薬ユーザーフィー法業績目標と手続きの再改定のセクションVIII(FDAの医薬品安全システムの強化と近代化)及びセクションIX(投薬過誤を削減するための商標名(ブランド名)の見直し)に記されている医薬品市販後安全対策業績目標を設定しました。計画は各実施項目、その背景、最近の実績を含むものとなっています。
2009年の活動とその後のフォローについて更新した活動実績の主なものを以下に掲げます。
 「計画-2009年更新版」は、第4次ユーザーフィー法医薬品安全対策の実績について、2009会計年度末までの進捗状況をまとめた初めての年間評価となっています。
「計画-2009年更新版」は、第4次ユーザーフィー法医薬品安全対策の実績について、2009会計年度末までの進捗状況をまとめた初めての年間評価となっています。
 情報提供依頼書の受理と精査の後、副作用自発報告情報の収集の効用を最大に発揮するための研究の提案依頼書が2009年7月17日に発出されました。FDAの副作用情報監視システム(サーベイランスシステム)が安全性に関連した規制当局の対応に寄与し貢献しているかの評価を実施しています。
情報提供依頼書の受理と精査の後、副作用自発報告情報の収集の効用を最大に発揮するための研究の提案依頼書が2009年7月17日に発出されました。FDAの副作用情報監視システム(サーベイランスシステム)が安全性に関連した規制当局の対応に寄与し貢献しているかの評価を実施しています。
 「大規模電子医療情報を用いた科学的に健全な薬剤疫学安全性研究を実施するためのガイダンスの作成」に関する公開のワークショップが2008年5月7日に開催されました。FDAの医薬品評価研究センター(CDER)と生物製品評価研究センター(CBER)の作業グループは、ワークショップでの討議とその後に寄せられた意見を踏まえ、ガイダンスの最終案作成作業に取り掛かっています。
「大規模電子医療情報を用いた科学的に健全な薬剤疫学安全性研究を実施するためのガイダンスの作成」に関する公開のワークショップが2008年5月7日に開催されました。FDAの医薬品評価研究センター(CDER)と生物製品評価研究センター(CBER)の作業グループは、ワークショップでの討議とその後に寄せられた意見を踏まえ、ガイダンスの最終案作成作業に取り掛かっています。
 目的とする市販後監視及び疫学的検討を行うため、CDER及びCBERのデータベースの取得及び利用の拡大を進めるために以下を実施しています。
目的とする市販後監視及び疫学的検討を行うため、CDER及びCBERのデータベースの取得及び利用の拡大を進めるために以下を実施しています。
1)CDERのメディケア・メディケイド・サービスセンター(CMS)とのプログラム
i) フルオロキノロンと急性アキレス腱断裂の危険性の研究
ii) プロポキシフェン及びその他の鎮痛薬の使用と骨折リスク、全死因死亡率、不整脈の関係
iii) 抗糖尿病薬(ロシグリタゾン、ピオグリタゾン)とフィブラート系薬剤(コレステロール降下剤:高脂血症改善薬)の2種類の併用と、高齢の2型糖尿病患者における心発作、脳卒中、死亡との相関関係
iv) エクセナチドのシタグリプチンとの比較(ともに2型糖尿病の治療の効能を持つ異なるタイプの薬剤)-高齢者における重篤な急性膵炎の研究
2)CDERの米国医療研究品質局(AHRQ)とのプログラム
i) 抗精神病薬(総合失調症治療薬)の安全性の比較評価
ii) TNFブロッカー(炎症状態を治療するために用いられる薬剤)の複数の安全性評価項目
iii) アセトアミノフェンの使用と安全性に関する知見の類型
iv) 小児入院患者の医薬品使用データと全国レベルでの小児入院患者医薬品使用の予測可能性
3)CDERの退役軍人健康庁(VHA)とのプログラム
i) HMG-CoA還元酵素阻害薬(スタチン)の使用と筋萎縮性側索硬化症(ALS)の危険性に関する研究-治験審査委員会(IRB)の承認を得て実施中。
ii) 抗精神病薬(総合失調症治療薬)の使用と死亡の危険性
iii) 抗精神病薬(総合失調症治療薬)の使用と遅発性ジスキネジア(異常運動)
iv) バレニクリン(禁煙補助製品として用いられる薬剤)と重度の精神障害-FDA,VHA、国防総省(DoD)との協同で実施。VHAの治験審査委員会(IRB)によるこの試験の許可は取得済み。DoDのIRBでは、医療記録の検証のため許可の手続き継続。
4)CDERの国防総省(DoD)とのプログラム
i) 疫学研究を通じた医薬品安全性情報(シグナル)の識別と確認のためのソフトウェアの強化
5)CDERの米国疾病管理予防センター (CDC)とのプログラム
i) 省庁間の合意に基づき、FDAはCDCに対し、全国電子傷害監視システム-共同医薬品有害事象監視(緊急医薬品情報データベース)をCDERの監視疫学部(OSE)の安全性評価スタッフと疫学研究者がより効率よく利用できるように、利用者に使いやすいクエ-リーツール(検索ツール)を開発するための資金を提供
ii) 全国電子傷害監視システム-共同医薬品有害事象監視(NEISS-CADES)あるいは全体的な米国電算機危害監視システム(NEISS)を用いてデータの抽出・収集を行うプロジェクトに2009年には、静脈内投与鉄剤とアレルギー反応、小児の偶発的なオピオイド誤飲(誤嚥)、アセトアミノフェンに関連した救急外来受診数を追加。
6)CDERの民間協力機関 (インジェニックス社、カイザー財団研究所、ヴァンダービルト大学、HMO医療保険研究ネットワーク)とのプログラム
i) 注意欠陥・多動性障害の治療に用いる薬剤と、それに関連すると思われる小児における重大な循環器系疾患の発現(心突然死、心発作、脳卒中)の危険性に関する多施設研究
ii) ビスフォスフォネート経口投与に関する2つの研究:1)ビスフォスフォネートとそれに関連した心房細動(AF)の危険性、2)ビスフォスフォネート使用者に見られた顎骨壊死の広がりとその危険因子に関する調査
iii) 新たな組み合わせによるホルモン避妊薬製品に関し、突然死、心発作、肺動脈塞栓症、深部静脈血栓症などの血栓関連事象の危険性を調査する多施設研究のためのデータの集積。
iv) 妊娠中の薬剤投与のリスク評価プログラム(MEPREP)と題する多施設プログラムにおいて2009会計年度は3種の実行可能性調査(予備調査)を実施中。妊娠期間中に使用された薬剤に関する研究と関連すると思われる新生児の有害事象とのデータの結合を開始。
7)CBERの取り組み
i) CMSとの協同でインフルエンザワクチン安全性プロジェクトを実施し、メディケアのデータを用いてインフルエンザワクチンの安全性について能動的に調査。CBERは多数のワクチンの中から、有害事象を迅速にモニタリングできる手法を開発しました。
ii) ワクチンの有害事象の準リアルタイム監視のため、CBERとCDCの間でのワクチンの安全性データ結合に関する省庁間合意。ワクチン安全性データリンク(VSD)は予防接種の安全性をモニターし、予防接種後の稀なあるいは重篤な有害事象の情報源となっています。FDAは、CDC予防接種安全部と8つのマネージドケア組織(MCOs)とが協力して実施している既存の活動に参加しました。FDAのスタッフは、CDC及びVSDの担当者と協力して2009-2010インフルエンザシーズンにおけるH1N1パンデミックワクチンでの事象に関する決定を行ってきました。また、FDAのスタッフはCDCと協同して、H1N1インフルエンザパンデミックワクチン、HPVワクチン、麻疹・風疹・おたふくかぜ・水痘(MMRV)ワクチン、13価結合型肺炎球菌ワクチン(PCV13)、水痘・帯状疱疹ワクチンの安全性の評価などを含め、特定の短期累積分析の優先課題の決定を行ってきました。
 リスクコミュニケーション諮問委員会では、FDA規制対象製品のリスクとベネフィットに関する一般とのコミュニケーションのプログラムと戦略を検証・評価し、FDAに対して医薬品の安全性についての一般の認識に関してアドバイスを行いました。
リスクコミュニケーション諮問委員会では、FDA規制対象製品のリスクとベネフィットに関する一般とのコミュニケーションのプログラムと戦略を検証・評価し、FDAに対して医薬品の安全性についての一般の認識に関してアドバイスを行いました。
 医薬品及び生物製品のリスク評価・軽減対策(REMS)の開発と実施に関する公開の会議を2010年7月27-28日に開催。FDAは広範囲の関係者からREMSプログラムの実施とガイダンス文書の作成に関する意見を聴取しました。
医薬品及び生物製品のリスク評価・軽減対策(REMS)の開発と実施に関する公開の会議を2010年7月27-28日に開催。FDAは広範囲の関係者からREMSプログラムの実施とガイダンス文書の作成に関する意見を聴取しました。
 新たなFDA有害事象リポーティングシステム(FAERS)の原型初案を開発。FAERSの目的は、医薬品の有害事象(AEs)の受動的(自発)報告の質、利用しやすさ、効率性を改善することにあります。FDA内のCDER,CBER,CDRH(医療機器・放射線保健センター)の間でのこのシステムは、有害事象を速やかに適切な評価者のもとに伝える仕組みを機能させるものです。
新たなFDA有害事象リポーティングシステム(FAERS)の原型初案を開発。FAERSの目的は、医薬品の有害事象(AEs)の受動的(自発)報告の質、利用しやすさ、効率性を改善することにあります。FDA内のCDER,CBER,CDRH(医療機器・放射線保健センター)の間でのこのシステムは、有害事象を速やかに適切な評価者のもとに伝える仕組みを機能させるものです。
 行政文書の保管、報告、追跡システム(DARRTS)に安全性に関係する事項についての通知の追跡を強化するための新たなカテゴリーを設定。FDAのスタッフは常時安全性モニタリングのひとつとして副作用報告システム(AERS)のデータベースを定期的に調査し、AERSのデータから重大なリスクの可能性が確認されると、その情報はCDERのDARRTS又はCBERの治療薬・血液安全性部門安全性情報追跡システム(SSTシステム)に安全性関係事項として入力されます。
行政文書の保管、報告、追跡システム(DARRTS)に安全性に関係する事項についての通知の追跡を強化するための新たなカテゴリーを設定。FDAのスタッフは常時安全性モニタリングのひとつとして副作用報告システム(AERS)のデータベースを定期的に調査し、AERSのデータから重大なリスクの可能性が確認されると、その情報はCDERのDARRTS又はCBERの治療薬・血液安全性部門安全性情報追跡システム(SSTシステム)に安全性関係事項として入力されます。
 CDERとCBERの薬剤疫学及び市販後監視の活動に関係する研修者は英国の一般診療研究データベース(GPRD)を利用しています。具体的例としては、
CDERとCBERの薬剤疫学及び市販後監視の活動に関係する研修者は英国の一般診療研究データベース(GPRD)を利用しています。具体的例としては、
i) 抗うつ薬の母体内暴露と新生児遷延性肺高血圧症
ii) 抗糖尿病薬とがんの危険性
iii) 新生児遷延性肺高血圧症の診療情報データベース内での新たなバリデーション手法
 FDAは、提案された商標名(ブランド名)が他の医薬品の名称とつづり字上の(スペリングの)又は音声学上の(音の)類似性を持つことで混乱をきたし、投薬過誤や不適切な量の投与をまねく危険性を増加させるような類似性があるかを見極めるためのコンピューター解析手法を開発しました。音声正字コンピューター分析システム(POCA)は商標名や医薬品名が見た目が似ているか、似たように聞こえるかを確認する自動分析のシステムです。ソースコードや関連技術文書の利用に関する通知は2009年に官報(FR)に掲載されました。
FDAは、提案された商標名(ブランド名)が他の医薬品の名称とつづり字上の(スペリングの)又は音声学上の(音の)類似性を持つことで混乱をきたし、投薬過誤や不適切な量の投与をまねく危険性を増加させるような類似性があるかを見極めるためのコンピューター解析手法を開発しました。音声正字コンピューター分析システム(POCA)は商標名や医薬品名が見た目が似ているか、似たように聞こえるかを確認する自動分析のシステムです。ソースコードや関連技術文書の利用に関する通知は2009年に官報(FR)に掲載されました。
 商標名を評価するための申請に必要な内容に関するガイダンスの最終版が2010年の2月に公表されました。
商標名を評価するための申請に必要な内容に関するガイダンスの最終版が2010年の2月に公表されました。
 CBERの標準業務手順書とポリシー(SOPP)及びCDERのポリシーと業務手順のマニュアル(MAPP)がそれぞれ2008年と2009年に公表されました。これらには、治験薬申請(INDs)、医薬品/生物製品の新薬申請(NDAs/BLAs)に含まれる商標名(ブランド名)についての内部手続きを含むものです。
CBERの標準業務手順書とポリシー(SOPP)及びCDERのポリシーと業務手順のマニュアル(MAPP)がそれぞれ2008年と2009年に公表されました。これらには、治験薬申請(INDs)、医薬品/生物製品の新薬申請(NDAs/BLAs)に含まれる商標名(ブランド名)についての内部手続きを含むものです。
 投薬過誤を削減するための名称設定、表示、包装の実務に関するガイダンスの開発についてのワークショップが2010年6月に開催されました。規制当局側、研究者、医薬品企業、医療関係機関の代表、医療関係者および一般からの参加者の間での討議内容は、ガイダンス案の作成に利用されています。
投薬過誤を削減するための名称設定、表示、包装の実務に関するガイダンスの開発についてのワークショップが2010年6月に開催されました。規制当局側、研究者、医薬品企業、医療関係機関の代表、医療関係者および一般からの参加者の間での討議内容は、ガイダンス案の作成に利用されています。
以上