
Overview
Importance of the Reliability of Application data
- Based on the application data submitted, risk-benefit balance of efficacy and safety is evaluated, and then the validity of indications/performance, dosage and administration, and precautions is reviewed.
- If the data submitted on approval application is unreliable, efficacy and safety in humans cannot be evaluated.
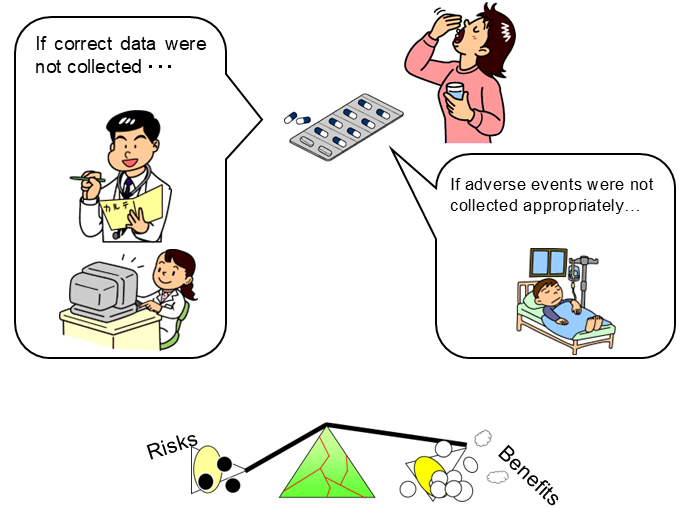
Laws & Regulations for Securing the Reliability of Application Data
<Pharmaceuticals and Medical Devices (PMD) Act(Note) Article 14, Paragraph 3>

(Note)Act: Act on Securing Quality, Efficacy and Safety of Products Including Pharmaceuticals and Medical Devices, Act No.145 of August 10,1960
What are “other data/documents” in the PMD Act Article 14 Para 3?
<Article 40 of the Enforcement Ordinance of the PMD Act>
The data/documents to be attached to the application form set forth in Article 38, Paragraph 1 or Article 46, Paragraph 1 pursuant to the provisions of Article 14, Paragraph 3 (…) of the Act (…) shall be the data/documents set forth in the relevant items.
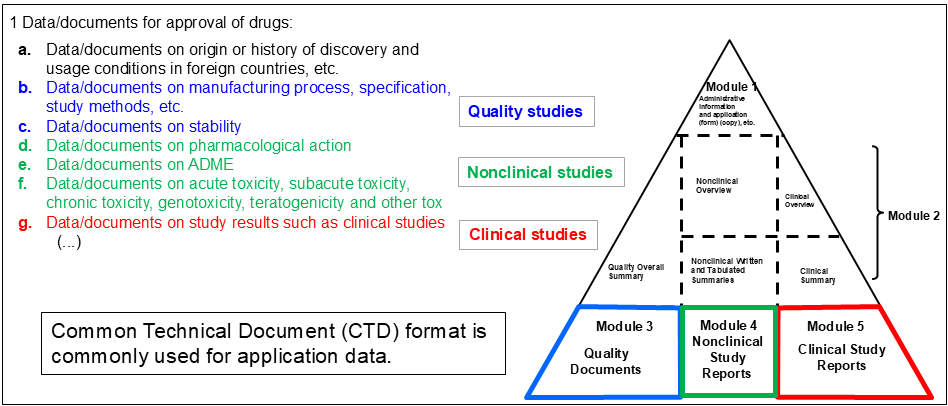
What are “the Standards specified by the MHLW Ordinance” in the PMD Act Article 14 Para 3?
<Article 43 of the Enforcement Ordinance of the PMD Act>

Series for Notification of inspection procedure

What’s the document-based inspection?
The following should be confirmed by inspecting raw data/documents for quality studies, non-clinical studies, and clinical studies in the application data.- Studies in the NDA dossier for new drugs have been appropriately conducted in an ethical and scientific manner in accordance with GLP/GCP and the study plan/protocol.
- Data/documents of the studies in the NDA dossier have been prepared appropriately and accurately in accordance with the standards provided in the Article 43 of the Enforcement Ordinance of the PMD Act, or so-called “Reliability Standards of Application Data” .
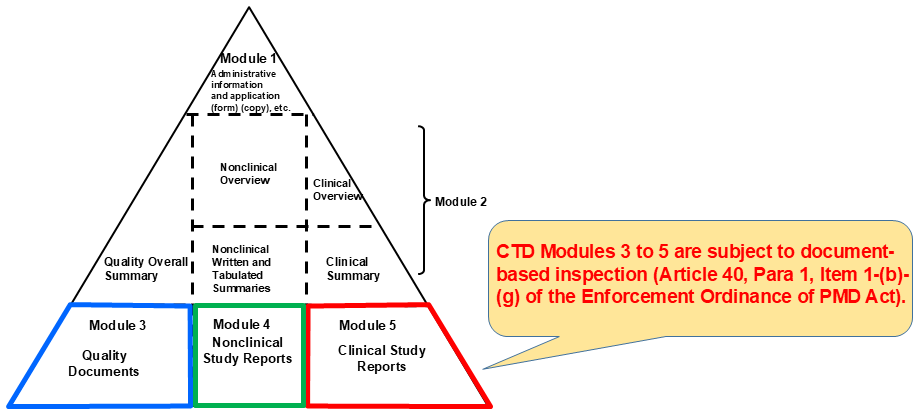
Scope of Inspection following the Reliability Standards of Application Data

- Unique to Japan
- Created for the purpose of recurrence prevention of drug-induced harm that have occurred in Japan in the past
- Common minimum requirements necessary to ensure the reliability of all data/documents (especially non-GxP studies) in application data
ALCOA+ vs Reliability Standards of Application Data
International principles for ensuring reliability
Attributable: Identification of the person performing the workLegible: Maintaining data readability and traceability
Contemporaneous: Recording data simultaneously with the work
Original: Maintaining original format
Accurate: Creating scientifically accurate data
Complete: Ensuring all data without missing data
Consistent: Maintaining consistency throughout the data lifecycle
Enduring: Ensuring maintenance of data for the future
Available: Data can be accessed as needed
Requirements in the Reliability Standards of Application Data for application data are in common with internationally recognized principles!!
Two ways to conduct the inspection to CTD M3(Quality studies)/M4(Non-clinical studies other than GLP studies)
- There are 2 ways to conduct a document-based inspection to CTD M3(Quality studies)/M4(Non-clinical studies other than GLP studies).
- PMDA decides the way in a critical consideration for the risk/importance of application data (e.g., application category).
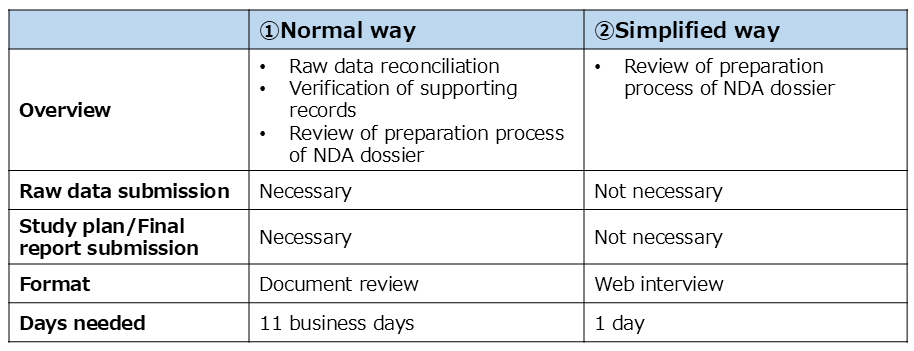
Document-based inspection(Normal way)
- Data/study(s) targeted for the inspection will be selected based on the risk/importance of data.
- The applicant will be required to submit the study plan and final report , raw data/documents of the data/study(s) selected.
- The inspection is conducted at desktop (self document review) in almost all cases.
- The main focus of the inspection is to reconciliate the raw data/original record with results in the CTD M3(Quality studies)/M4(Non-clinical studies other than GLP studies).
- In addition, supporting records and QC/QA process of CTDM3(Quality studies)/M4(Non-clinical studies other than GLP studies) will be reviewed in the inspection.
Timelines
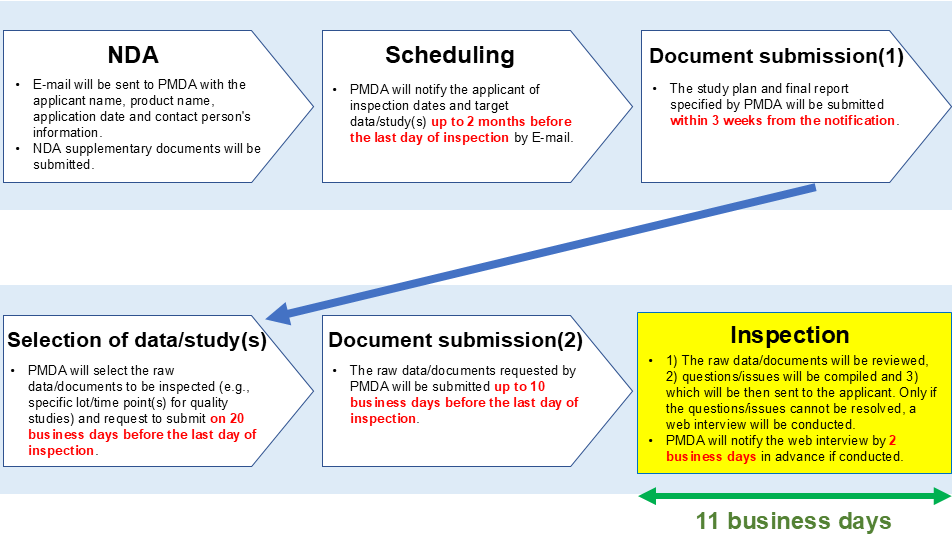
Data/documents to be submitted

Types of raw data/documents to be submitted
- A typical inspection will require the submission of raw data and other relevant records which are directly linked to the test results in the CTD.
- There are two types of data/documents for Normal way: "Data/documents to be submitted as default" and "Data/documents to be submitted as needed." (However, there may be cases where the volume of data/documents is reduced by the conditions and scope specified) For "Data/documents to be submitted as needed", inspectors will request to submit during the inspection period (or before) only if they determine to be necessary.

Important Notice for raw data/document submission
- Please coordinate with the inspector individually if the raw data/documents to be submitted are in a file format that cannot be read without specific software (instead, may ask presenting with a screenshot or sharing the screen at a web interview).
- Please make the orientation of the documents consistent, as there have been cases where the orientation of submitted PDF files has not been consistent when storing submitted data/documents in a cloud or other system.
- The “Description of raw data/documents” should include (1) Document showing the folder structure, (2) explanation of the flow of data, and (3) other information useful for efficient inspection (see the next item).
- Please submit the “Description of raw data/documents” in the same method as raw data/documents (i.e., the cloud system or the Gateway System)
- Raw data/documents to be submitted will be selected as follows. It may be further narrowed in consideration of data volume.
Quality studies: Lot number, measurement time point, etc. (could be selected by measurement items)
Non-clinical studies: All raw data/documents, but could be selected by measurement items, treatment groups, etc.
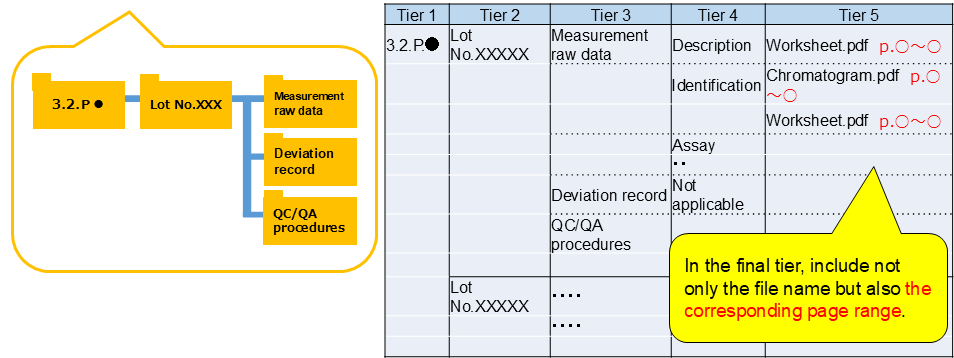
Description of raw data/documents
Please submit ”Description of raw data/documents’’ including the following imformation (1)-(3), otherwise it might lead to an increase in the number of questions or an extension of the inspection period.(1)Document showing the folder structure (explains in detail the structure of raw data/documents submitted)
- Submit a document showing the folder structure with raw data/documents and should also send it by e-mail.
- Organize the data/documents per data/document type listed in the previous table.
- Name the data/documents so that the inspectors make it possible to guess the contents.
- Create subfolders for each category of documents.
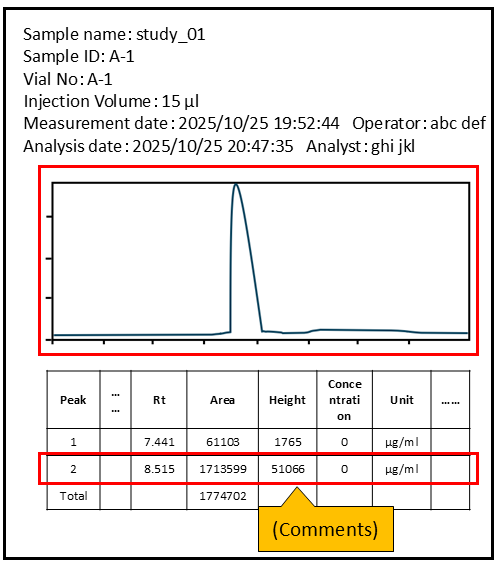
(2) Explanation of data flow
Create a certified copy of the raw data/documents and use it to address the following items- The flow of data (Lot number, measurement results, Chromatogram, Calculated results, etc.) in red boxes or highlight them.
- Comments if necessary.
(3) Other information useful for efficient inspection
e.g.) If the raw data/documents were recorded in a language other than Japanese and English- Although do NOT have to translate all of the contents, translate (in Japanese or English) enough for inspectors to be able to follow the flow of data (study items, measurement results, etc.) at least.
- Translation of specific parts of raw data/documents may be requested during the inspection period.
How to submit raw data/documents
(1)On-line Storage systems such as Cloud systems
- The applicant shall provide the inspectors permission to (read-only) access to the system.
- The following systems are not able to be used under the PMDA’s network environment.
e.g.) Systems requiring application installation
Systems requiring change of security settings
Systems requiring file download
- There are some requirements for word search, word copying, etc.
- A connection test will be performed prior to uploading the raw data/documents.
(2) Electronic Study Data Submission System (“Gateway System”)
URL:https://esg.pmda.go.jp/Ssk/comn001p01.init [Japanese only]
For details, please refer to the following official notification!
Procedure for Remote Inspection as a Part of Compliance Inspection on Drugs and Regenerative Medical Products[197KB]
Document-based inspection(Simplified way)
- The main focus of the inspection is to confirm the preparation process for the NDA dossier by Web interview.
- Therefore, an applicant is NOT required to submit the study plan, final report and raw data/documents.
Timelines

Items to be explained during Web interview
(1)Background information of NDA(2)Preparation process for NDA dossier
- QC/QA and approval process (including person/entity responsible) from the raw data to CTD (NDA dossier) will be clearly identified using a flow chart.
- The records of approval for each step will be presented.
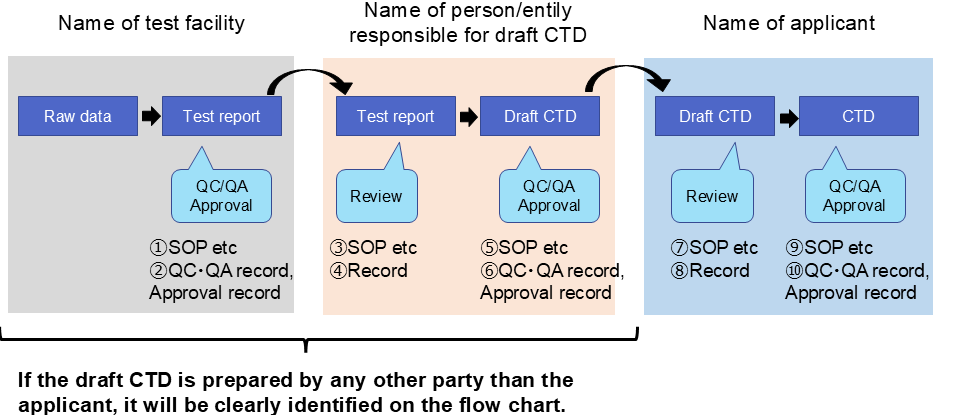
- Applicant’s procedure to confirm the completion of the preparation process by any other party will be explained if the above-mentioned step2 was not performed by the applicant.
Reference materials
Self-inspection checklist Q&A for quality and non-clinical studies
The checklist was revised to restore its original purpose as a reference for applicants' self-inspections and is available on the Japan Pharmaceutical Manufacturers Association (JPMA) website.Regulations and Notifications
Drugs
- Inspection guidelines (MHLW PSEHB/PECBD Notification No.0703-1, dated Jul 3, 2023)[782.10KB] [Japanese only]
- Inspection procedures (PMDA Notification No.2771, dated Jul 3, 2023)[366.86KB][Japanese only]
- Procedure for Remote Inspection as a Part of Compliance Inspection on Drugs and Regenerative Medical Products(PMDA/CPE Notification No. 325, dated July 3, 2023 )[197KB]
- Supplement of notification of remote inspection[531KB][Japanese only]
Regenerative Medical Products
- Inspection guidelines (MHLW PSEHB/PECBD Notification No.0703-1, dated Jul 3, 2023)[365.51KB] [Japanese only]
- Inspection procedures (PMDA Notification No.2772, dated Jul 3, 2023)[404.72KB][Japanese only]
- Procedure for Remote Inspection as a Part of Compliance Inspection on Drugs and Regenerative Medical Products(PMDA/CPE Notification No. 325, dated July 3, 2023 )[197KB]
- Supplement of notification of remote inspection[531KB][Japanese only]
Related Information
- Document-based Inspection to CTD M3/M4 data[1.21MB](December 2025)