
品質・非臨床試験に係る適合性書面調査の概要
「信頼性の基準」の位置付け
- 薬機法 第14条第3項では、承認申請に係る資料すなわち申請資料は、厚生労働省令に従って収集・作成されている必要がある旨が規定されています。なお、本項は医薬品を例とした説明であり、再生医療等製品は参照する法令・通知等が異なりますが、内容は医薬品と同様です。
<薬機法 第14条第3項> (注)医薬品、医療機器等の品質、有効性及び安全性の確保等に関する法律

- 薬機法 第14条第3項に規定する「添付資料」には臨床試験の成績以外にどのような資料が必要か具体的に規定しているのが、薬機法施行規則になります。トの臨床試験の他に、ロハに該当する品質試験、ニホヘに該当する非臨床試験の成績を添付することが求められています。医薬品の承認申請資料を提出する際には、ICHで合意されたCommon Technical Document(CTD)の形式で提出されることが一般的であり、このCTD形式に基づくと、品質試験は第3部、非臨床試験は第4部、そして臨床試験は第5部に纏められることになります。
<薬機法施行規則 第40条>
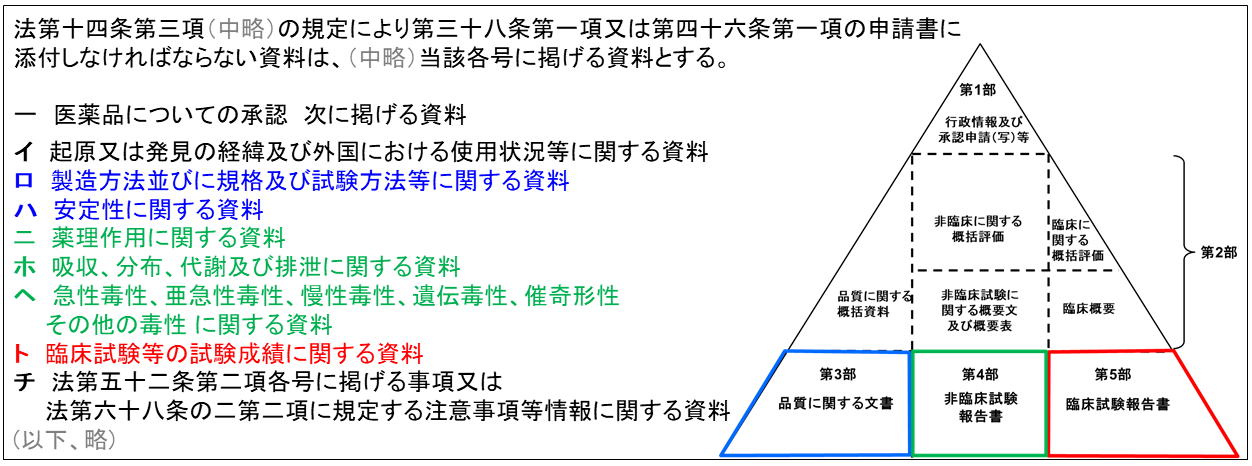
- 薬機法施行規則 第43条において、「申請資料はGLP、GCP、GPSP省令の他、次に掲げるところにより、収集され、かつ、作成されたものでなければならない」旨が記載されており、正確性・網羅性・保存性に関する3つの事項が掲げられています。信頼性の基準とは、当該3つの事項を遵守することであり、承認申請に添付される全ての資料に対して試験成績の信頼性確保のために必要最小限求められる事項です。本邦において過去に発生した薬害を教訓として創設された、主要国の中でも本邦独自の基準ですが、その考え方自体は国際的な原則のALCOA+とも共通しています。
<薬機法施行規則 第43条>

「適合性書面調査」の位置付け
- 薬機法第14条第6項には「承認申請資料が厚生労働省令で定める基準に適合しているかどうかを書面若しくは実地の調査によって確認する」旨が規定されているとともに、同第14条の2の3第1項には「この調査を機構に行わせることができる」旨が規定されています。当該記載に基づき、承認申請資料のGxP・信頼性の基準への適合性の確認・調査はPMDAが担っていますが、実際に調査を行う際の手順・手続きは、厚生労働省医薬品審査管理課長通知である調査実施要領通知、PMDA理事長通知である調査実施手続き通知に明文化され、これらに従って実施されます。

- 2020年より、クラウド等システムやweb会議システム等を通じて遠隔的に根拠資料を確認する調査である「リモート調査」を開始したことに伴い、上述した通知等に加え、PMDA審査センター長通知であるリモート調査通知を発出しています。これは、調査手法の一つとなったリモート調査を実施する場合の取り扱いを明文化したものです。さらに、リモート調査実施に関する詳細な運用を補足する資料として、リモート調査通知の第5項に規定する資料を掲載しており、リモート調査を運用していく中で実務的で詳細な事項が記載されています。

(注)上記通知及び補足資料は、「6.関連通知等」にリンクを掲載しています。
品質・非臨床試験に係る適合性書面調査の概要
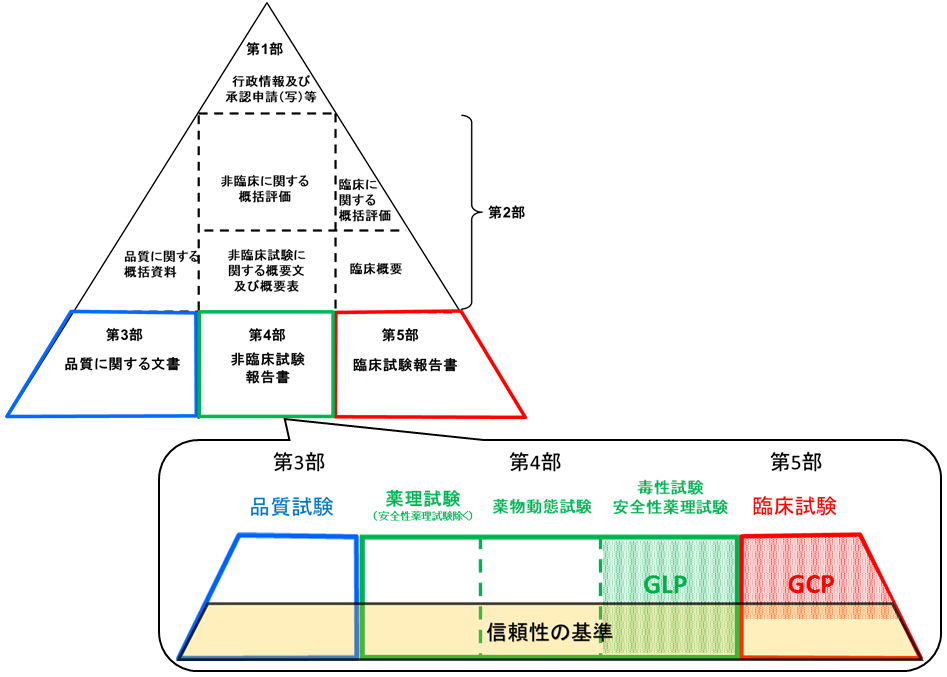
- 信頼性の基準は、CTD第3部から第5部の資料について広くカバーしています。
- 臨床試験及び安全性に係る非臨床試験は信頼性の基準に加えて、それぞれGCP及びGLPへの適合が求められます。一方で、GxPが適用されない品質試験、非臨床試験のうち薬理試験(安全性薬理試験を除く)及び薬物動態試験は信頼性の基準への適合のみが求められます。
- これらの申請資料について、品質・非臨床試験に係る適合性書面調査を実施し、信頼性の基準に基づき作成されていることをリスクに応じた適切な手法で確認しています。
- 品質・非臨床試験に係る適合性書面調査の実施方法には、(1)標準的な調査、(2)経緯確認調査、(3)中等度変更事項調査(試行中)の3種類があり、原則として一律リモート調査にて実施します(緊急案件の場合等は、訪問調査や機構への持ち込み調査を実施する可能性があります)。 各実施方法の概要は次項をご参照ください。
調査の実施方法及びスケジュール
標準的な調査
- 標準的な調査では、試験結果に係る生データ、温湿度記録・逸脱記録等の関連記録、申請資料作成に係るQC・QA手順を確認します。そのため、調査直前提出資料の提出及びリモート調査用資料の提示が必要となります。
- 標準的な調査における品質試験及び非臨床試験の調査対象資料は、臨床試験も含めた承認申請資料全体から、そのリスクと資料の重要度に応じて選定しています。
- 標準的な調査の流れは下図のとおりです。
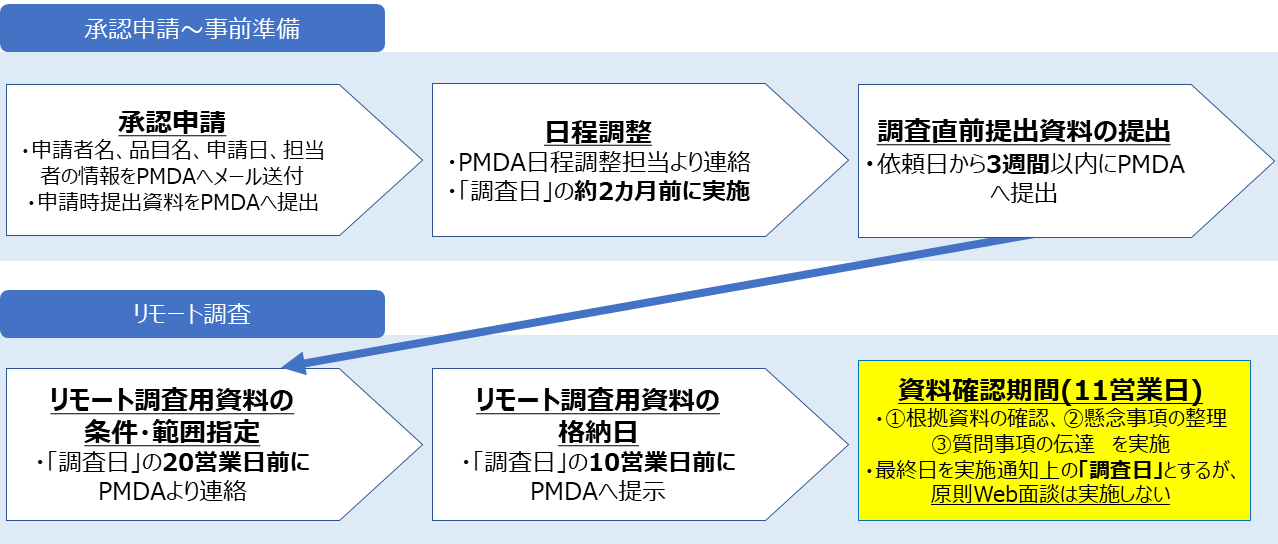
- 品質・非臨床試験に係る適合性書面調査の実施通知上の「調査日」は資料確認期間の最終日としておりますが、原則、品質・非臨床試験に係るWeb面談は実施しません。ただし、資料確認期間中に懸念事項が解消せず、議論する必要があると判断した場合は、2営業日前までにWeb面談の設定を依頼します。
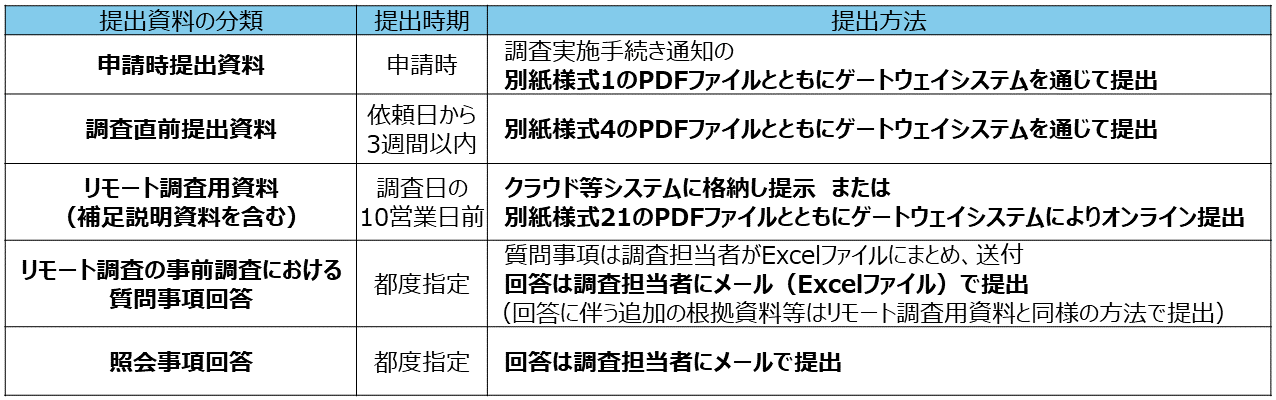
- 標準的な調査で提出が必要な資料は以下のとおりです。なお、下表のうち、リモート調査用資料については「3.標準的な調査におけるリモート調査用資料」に、補足説明資料については「4.標準的な調査における補足説明資料」に詳細を記載しています。
- 別紙様式は調査実施手続き通知(「6.関連通知等」にリンクを掲載)に添付されており、以下のPMDAサイトからもダウンロードできます。
https://www.pmda.go.jp/PmdaSearch/youshikiDownload/
- 臨床試験の適合性書面調査がある場合、手続きの一部を臨床試験と併せて実施することがあります。
経緯確認調査
- 経緯確認調査とは、標準的な調査と異なり、根拠資料の確認を省略し、申請に至る経緯を確認するものです。
- 申請区分等を勘案したリスクに応じて実施します。例えば、医療用医薬品の申請区分(10)に該当する申請が経緯確認調査の対象となる可能性があります。
- 経緯確認調査では、「申請資料の作成プロセス」にフォーカスして、そのQC及びQA手順をWeb面談により確認します。そのため、調査直前提出資料の提出及びリモート調査用資料の提示は不要です。
- 経緯確認調査の流れは下図のとおりです。

- 調査当日にWeb会議システムを通じて画面共有により下記(1)から(3)について説明していただきます。
(1)申請の概要
変更事項に関する概要を変更点が分かるように説明すること。

(2)申請資料作成のプロセス
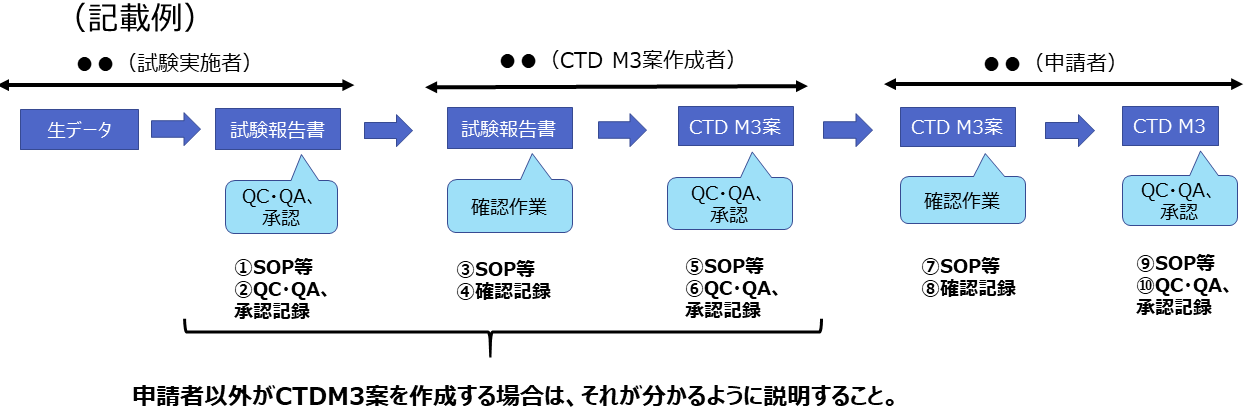
生データから試験報告書、CTD M3(品質に関する文書)の作成に至るプロセスをフロー図等で示すこと。
実施主体(試験実施者、CTD M3案作成者、申請者等)を明確にしたうえで説明すること。
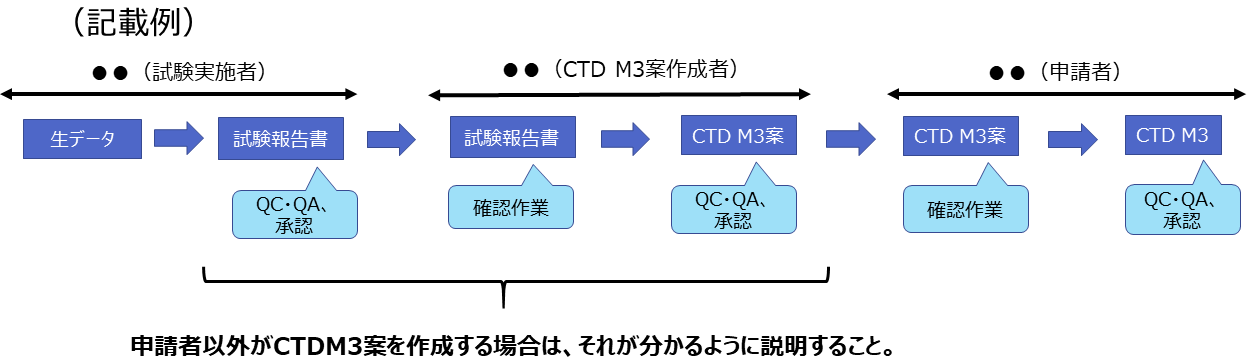
(3)上記(2)のプロセスが適切に実施されていることを申請者が確認する手順及び確認した内容
実際の作業手順(SOP・手順の概要等)や発生した記録(QC・QA・承認記録・確認記録等)を具体的に示し、説明すること。
中等度変更事項調査(試行中)
- 医薬品の製造方法等の変更手続は従来、「一部変更承認申請」と「軽微変更届出」により行ってきましたが、「一部変更承認申請」のうち、よりリスクの低いもの(規格及び試験方法や有効期間の変更等)は迅速に審査を行うため、試行的に「中等度変更事項」という新たな区分を設定して変更手続きを行っており、その区分に対する適合性書面調査として「中等度変更事項調査」を実施しています。
- 中等度変更事項調査は、経緯確認調査をより簡略化した方法で実施しています。詳細は下記ファイルをご確認ください。
標準的な調査におけるリモート調査用資料
リモート調査用資料の概要
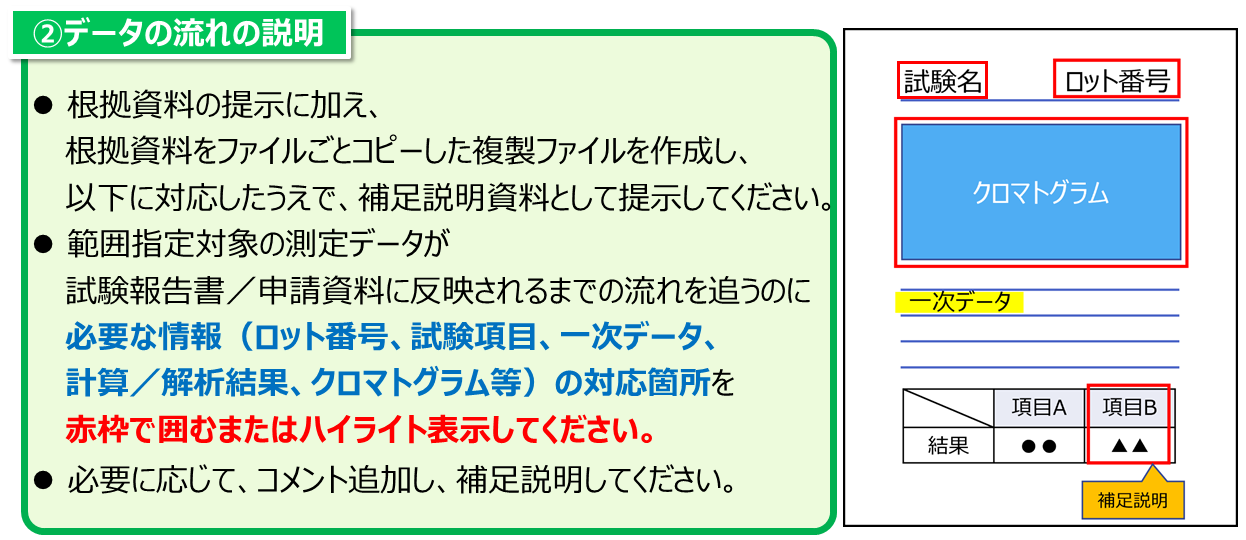
- 標準的な調査では、リモート調査用資料として、試験結果に直結する生データやその他の関連する記録の提示が必要となります。
- リモート調査用資料には、「選定された試験において原則的に提示を求める資料」と「必要に応じて提示を依頼する資料」があります(ただし、資料の条件・範囲を指定する場合あり)。「必要に応じて提示を依頼する資料」は、提示が必要と判断された場合のみ、資料確認期間中(若しくはそれ以前)に調査員より提示の依頼があります。

- 特定のソフトでないと見読できないファイル形式の場合は、提示方法について個別にご相談ください(スクリーンショットでの提示や、Web面談時の画面共有を検討)。
- 提示されたPDFファイルの方向が縦横統一されていない事例が散見されます。提示資料をクラウド等システムに格納する際には、資料の向きを確認し、統一してください。
- 生データは該当する箇所を切り抜いた画像ではなく、ページ全体を提示してください。
- 補足説明資料については、「4.標準的な調査における補足説明資料」にて詳細を説明しています。
リモート調査用資料の提示範囲
- 生データは、以下のとおり提示範囲を指定します。データ分量等を考慮し、さらに限定する場合もあります。
品質試験:ロット番号、測定時点等 (測定項目等を限定する場合あり)
非臨床試験:(指示がない限り)全ての生データ (測定項目、投与群等を限定する場合あり)
標準的な調査における補足説明資料
補足説明資料の目的
- 標準的な調査では、リモート調査用資料に加え、補足説明資料を提示いただく必要があります。
- 補足説明資料は、リモート調査通知(令和5年7月3日付け薬機審長発第325号「医薬品及び再生医療等製品の適合性調査におけるリモート調査の実施方法について」)の第3項(2)②に規定されている資料であり、原則、リモート調査の対象となる全ての品目において提示を求める資料となります。
- しかし、現在までの適合性書面調査において、補足説明資料が提示されない、または提示されていても情報が不足している事例が散見されています。補足説明資料の内容が不十分な場合、事前質問事項の増加や調査期間の延長につながる恐れがあります。効率的な調査実施のため、補足説明資料の作成をお願いします。
補足説明資料の提示方法
- 補足説明資料は、リモート調査用資料と同様の方法(クラウド等システムへ格納、ゲートウェイシステムによるオンライン提出等)で提示してください。
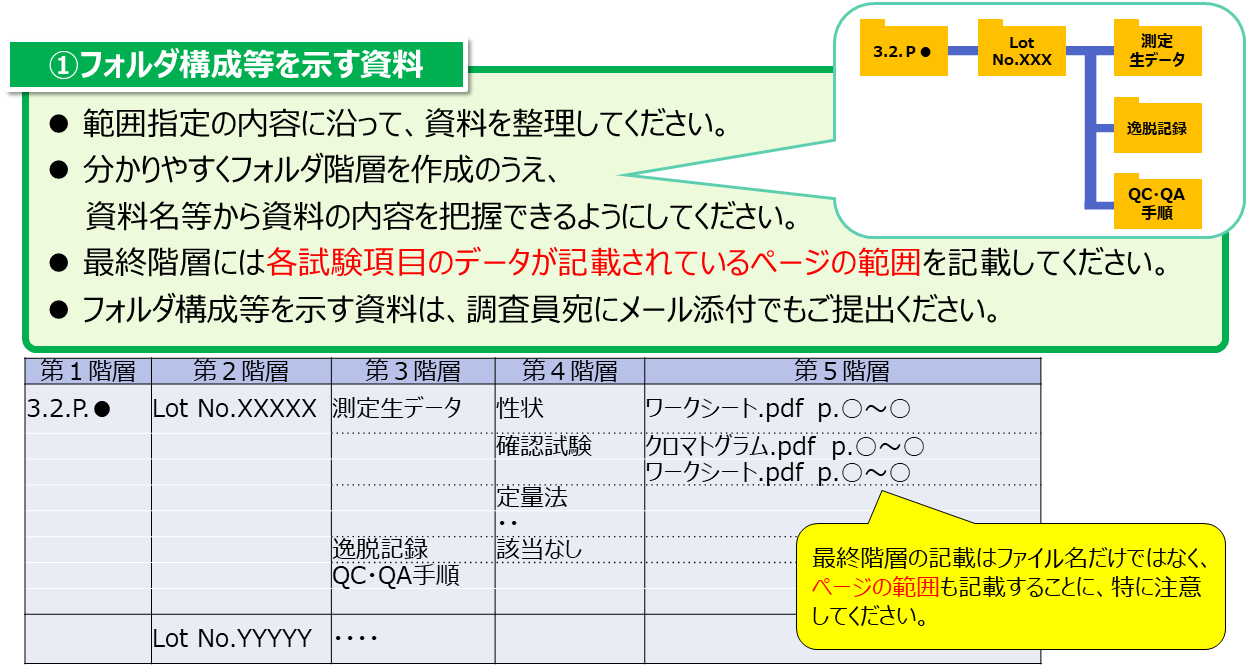
- フォルダ構成等を示す資料(提示した根拠資料の構成の詳細)は、調査員宛にメール添付でも提出してください。
補足説明資料の作成
- 品質試験・非臨床試験の調査の場合、補足説明資料には以下(1)から(3)の情報を含めてください。
なお、フォルダ構成等を示す資料(提示した根拠資料の構成の詳細)のテンプレートは以下のとおりです。
フォルダ構成等を示す資料(テンプレート)[10.5KB]

品質・非臨床試験に係る適合性書面調査の留意事項
申請時のメール送付
- 申請日から5営業日以内に、機構(メールアドレス:gcp_inspection[at]pmda.go.jp)宛に、「申請者名」、「品目名」、「申請日」及び「適合性調査の窓口となる担当者」の情報を記載したメールを送付してください。
- なお、適合性書面調査の申請のみが行われている品目(医薬品GCP実地調査の申請が行われていない品目)であり、かつ審査予定事前面談が実施されていない場合は、機構(メールアドレス:tekigousei-shomen[at]pmda.go.jp)宛に送付してください。
(注)迷惑メール防止対策をしているため、送信の際は[at]を半角のアットマークに置き換えてください。
添付資料一覧(CTD M1.12)に関する留意事項
- 標準的な調査においては、CTD M1.12添付資料一覧の記載に基づき、M3、M4から調査対象資料を選定しており、資料が細分化されている場合、原則細分化された資料から選定しています。
- 細分化する場合、各資料に特有のCTD番号の枝番を付してください。また、対応する各資料本体は、可能な限りM1.12のCTD番号と一致する枝番・タイトルを明示した個別ファイルとしてください。

- 資料のタイトルにCTD番号は記載しないでください。

- 一部の企業において、CTD M1.12添付資料一覧のM3(品質に関する文書)関連の記載粒度が粗い場合があり、調査対象資料の特定に支障が生じています。その場合、調査用資料として品質部分の記載を詳細にした添付資料一覧の再提出が必要となるため、事前に確認をお願いします。
- CTD M1.12添付資料一覧の記載粒度についてはICH-M4Qを参考に申請資料の作成をお願いします。

ゲートウェイシステムを通じた資料のオンライン提出方法
- 申請時提出資料、調査直前提出資料及びリモート調査用資料の提示には、Zipファイル(パスワード付きファイルは不可)及び「別紙様式1、4、13、16及び21」はPDF形式のファイルをご準備ください。
(注1)別紙様式は、Zipファイルに含めないでください。
(注2)資料の差し換え/追加提出の場合も別紙様式を添付してください。 - Zipファイル名は、「品目名(略称)_資料名(別紙様式/申請時提出資料/調査直前提出資料/リモート調査用資料等)_(通し番号)_提出日(yyyymmdd)」としてください。
(注)現時点で通し番号にルールは設けていません。各社で管理しやすい形で付番してください。 - フォルダが階層化されている場合、丸ごとZip化すればフォルダ階層を維持したまま提出が可能です。なお、Zipファイルのサイズが1GBを超えないようにしてください。
- ゲートウェイ提出時の手続き分類の選択について、分類1及び2は申請内容に合わせて選択し、分類3は「XXX:申請時提出資料、調査直前提出資料、リモート調査用資料」を選択してください。分類3の「XXX:機構が指示した資料」は、調査員がその区分を指示した場合のみ使用可能です(それ以外の場合で使用されても資料提出とはみなしません)。
- 提出名称は「品目名(略称)_申請日(yyyymmdd申請)」と記載してください。また、1つの承認申請で複数回提出する場合でも初回と同じ提出名称を入力してください。
- ゲートウェイ受付番号も提出名称と同様に1つの承認申請で1つのゲートウェイ受付番号を通しで使用してください。
- ゲートウェイシステムに関するその他留意事項は以下ファイルをご確認ください。
ゲートウェイシステムを通じた医薬品及び再生医療等製品の適合性書面調査、GCP/GPSP実地調査の資料の提出方法について[1.08MB](2023年7月31日更新)
クラウド等システムに関する留意事項
調査担当者は、機構のネットワーク環境を通じてクラウド等システムに接続します。機構のネットワーク環境で接続又は利用できないクラウド等システム(アプリケーションのインストールやセキュリティ設定の変更が必要なシステム、ファイルのダウンロードが必要なシステム等)である場合には、他のクラウド等システムを利用する等、根拠資料(電磁的記録)の提示方法を変更してください。クラウド等システムは、リモート調査通知4. に記載がありますが、特に重要な点を以下に抜粋します。
接続テスト
- システムに対するアクセス権限を設定する際、原則、次の機能を使用できる閲覧権限のみのアカウントを発行してください。
文字検索(紙資料をスキャンして作成したファイルを除く)
文字コピー(紙資料をスキャンして作成したファイルを除く)
Excelフィルター設定(事前にフィルター機能を設定、格納いただくことでも可)
- クラウド等システムの接続テスト時には、上記機能のテストが可能なファイルを格納してください。
- クラウドアクセスのための二段階認証は携帯電話番号(SMSや音声通話)又はメールアドレスをご利用ください。スマートフォンにダウンロードが必要となるアプリケーションを用いた二段階認証は使用できません。
調査員が使用するアカウントの管理
- 調査結果が通知されるまでクラウド等システムに保存した根拠資料及び調査担当者のアカウントを保持してください。
- 結果通知書受領後は、セキュリティの観点から速やかに、当該調査資料への調査担当者アクセス制限及びアカウントの削除(または無効化)を実施してください。
- ダウンロードの可否に関わらず、不正アクセス等の防止措置を講じてください。
- なお、不正アクセス防止のためにログを活用いただくことは全く問題ございませんが、セキュリティ確保以外での目的でのログの確認はご遠慮ください。
調査員が使用するアカウントの権限
- 誤操作防止の観点から、可能な限り、編集及びダウンロード等の権限を有するアカウントの発行を避けてください。申請者等の責任において、これらの権限を有するアカウントを発行する場合には、あらかじめ調査担当者から許可を得るとともに、留意すべき事項を連絡してください。
- 上記の場合、調査担当者が誤ってダウンロードした際には、削除等により対応することとなります。調査員が誤ってダウンロードする可能性を許容できない場合は、当該システムはご利用いただけません。
- また、ダウンロード履歴等からすべての資料がダウンロードされている等、不正アクセスが疑われると判断された場合には、当該システムを利用停止いただいたうえで、調査担当者にお問い合わせいただくこととなります。
クラウド等システムのフォルダ名
- クラウドのフォルダ名には品目を特定できる情報【品目名、申請者名】を記載してください。
(記載例)「リモート調査用(XX錠、YYYY会社)
- アクセス先URLをメールにコピー&ペーストして連絡いただくことがありますが、社外からアクセスできないURLが案内されるケースがあります。原則、クラウド等システムから生成される正式な案内メールの送付をお願いします。
Web会議システムに関する留意事項
- 事前打合せ、Web面談等の参加者の氏名・所属について、調査担当者にメールで事前に連絡してください。事前連絡されていない者が参加する必要が生じた場合には、調査担当者に氏名・所属を伝えた上で参加してください。
- 発言者は、カメラ機能をオンにしてください。また、発言者以外は音声機能をミュート設定にしてください。
- 記録作成等の目的のために、Web面談の内容を録音する場合には、調査担当者に事前にご連絡ください。
- 録音したデータの利用は、適合性調査の目的の範囲内であって社内に限定することとし、外部利用 (学会における発表等)又はインターネット等を通じて外部に漏洩しないようにしてください。また、調査結果が通知された後、速やかに消去してください。なお、対面助言の運用にあわせて録画及びWeb会議システムに付随する録音・文字起こし機能の利用は許可しないこととしますのであらかじめご承知おきください。
英語版の説明資料等
- 品質・非臨床試験に係る適合性書面調査について、海外向けに作成した英語版の資料は以下のとおりです。
Document-based Inspection to CTD M3/M4 data[1.21MB](December 2025)
(注)以前掲載していた「Compliance Inspection under PMD Act」(February 2023)については情報が古くなったため廃止とし、最新の情報を踏まえて新規に本資料を作成しました。
関連通知等
掲載されている各種関連通知のうち、品質・非臨床に係る適合性書面調査に関連するものを以下に抜粋します。
6.1.GCP実施調査及び適合性書面調査 実施要領
- 新医薬品の承認申請資料適合性書面調査、医薬品のGCP実地調査及び医薬品のGPSP実地調査等に係る実施要領について[782.10KB]
(2023年(令和5年)7月3日 薬生薬審発0703第1号)
(2023年(令和5年)7月3日 薬生機審発0703第1号)
6.2.GCP実施調査及び適合性書面調査 実施手続き
(2023年(令和5年)7月3日 薬機発第2771号)
(2023年(令和5年)7月3日 薬機発第2772号)
6.3.リモート調査通知
(2023年(令和5年)7月3日 薬機審長発第325号)
6.4.リモート調査通知 第5項に規定する資料
(2023年(令和5年)7月14日更新)
(2023年(令和5年)7月14日更新)
(2023年(令和5年)7月14日更新)
6.5.その他
よくある質問
参考資料
品質試験・非臨床試験に係る自主点検用チェックリスト
対象:品質試験、非臨床試験(薬理試験、薬物動態試験)申請者による自主点検の際に参考とするものという本来の主旨に戻り、申請者側で作成するチェックリストとして改定を実施しました。
改定に伴い、PMDA HPから日本製薬工業協会(JPMA)のHP上での公開に変更されました。
- 品質試験チェックリスト、Checklist for quality testings(English)
- 非臨床試験チェックリスト
- 非臨床試験に係る自主点検用チェックリスト Q&A
- Noclinical-checklist&QA(English)
信頼性ニュース号外(信号)
関連学会等で話題になっている事項や信頼性調査において改善を促した事項のうち、企業、医療機関及び受託機関等の調査対象者に対して早期に周知・注意喚起することで非臨床試験や治験等の環境整備に特に有用(注)と考えられる事例について、「信頼性ニュース号外」(略称:「信号」)として公表しています。そのうち、品質・非臨床試験に係るものについて、抜粋して以下に掲載します。
(注)個々の調査対象者等に限定される事例ではなく、試験・データの信頼性への影響が大きく関係者の参考となるもの。