
PMDAに寄せられた申請電子データに関する問合せをQ&A形式でまとめました。
申請電子データに関わる疑問点、不明点を解決する手段として、ご利用ください。
本ページのQ&Aはこちら[819KB]のPDFファイルにもまとめております。
また、本ページのQ&Aを関連する事項毎にまとめたPDFファイルもこちら[1.10MB]にありますので、併せてご利用ください。
申請電子データに関する審査・相談制度についての質問
Q1-1:(削除)
Q1-1-1:(削除)
Q1-2:ゲートウェイ申請において、承認申請書(FD申請データを含む。)の鑑、「提出年月日」欄及びeCTDカバーレターにおける「申請日」欄には申請を予定している日を、eCTDカバーレターの「提出日」欄にはゲートウェイシステムにおいて電子ファイルの提出を予定する日を記載するのでしょうか。
A: そのとおりです。「申請日」欄及び「提出日」欄等に記載する日付の考え方は従来どおりです。また、ゲートウェイシステムを利用してFD申請データの差換え版やeCTD改訂版を提出する場合は、送信を開始する予定日を記載してください。
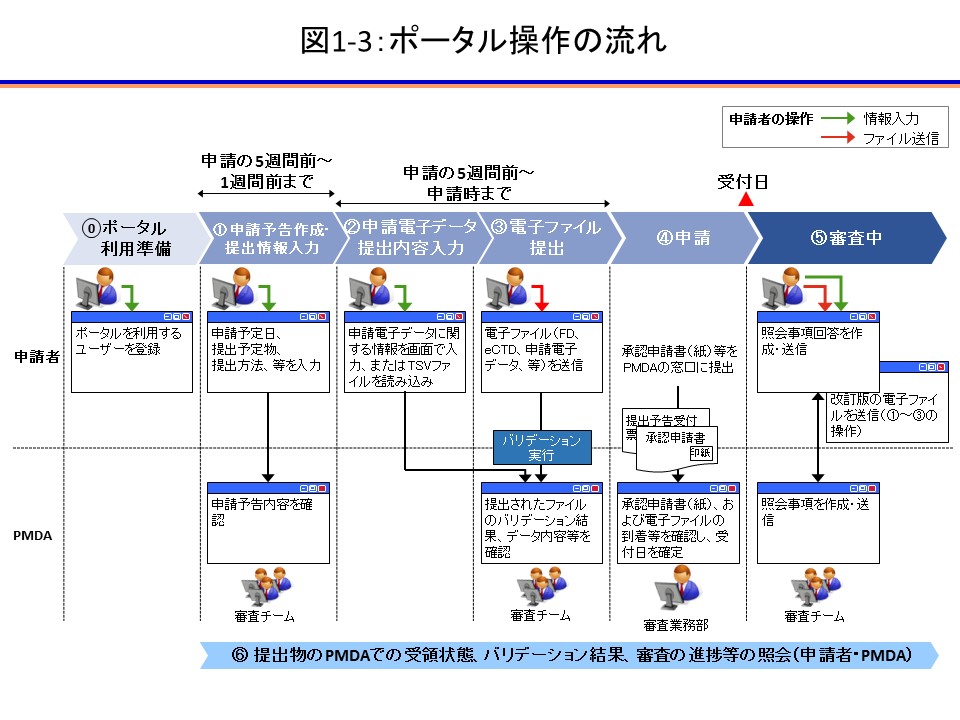
Q1-3:ゲートウェイシステムを利用する場合、承認申請の流れと共に、申請者はいつ何をすべきなのかを簡単に説明してください。
A: ゲートウェイシステムを利用する承認申請は下記の流れとなります。
【 】内にその作業が可能又は推奨する期間を記載します。
a)個人の電子証明書を用意し、ゲートウェイシステムのユーザー登録を行います。(図1-3 ⓪)
【申請予定日より1ヶ月程度前を推奨】
b)ゲートウェイシステムから承認申請の概要や提出予定物、申請予定日をPMDAに予告します。提出予告により、eCTD受付番号を取得することができます。(図1-3 ①)
【申請予定日の5週間前から1週間前】
※申請予定日の1週間前を切ると申請を予告できなくなりますのでご注意ください
c)ゲートウェイシステムから電子ファイルをPMDAに提出します。申請電子データに関する情報は、画面上で入力するか、別途作成したTSVファイルを読み込むことで必要な情報を登録します。(図1-3 ①~③)
【b)の申請日を予告してから、d)の申請までの間】
d)提出を予定する全ての電子ファイルのウイルスチェックが完了し、FD申請データのバリデーションに問題がないことが確認されれば、承認申請が可能となります。申請予告受付票を印刷し、承認申請書とともにPMDA受付窓口に提出します。(図1-3 ④)
Q1-4:(削除)
Q1-5:治験相談、新医薬品の申請電子データの提出に係る各相談及び新医薬品承認審査予定事前面談について、その取り扱う内容と留意点について、教えてください。
A: 治験相談、新医薬品の申請電子データの提出に係る各相談及び新医薬品承認審査予定事前面談について、その取り扱う内容と留意点は、以下の表のとおりです。懸念事項について、いずれの相談区分で相談すべきか判断に迷う場合は、適宜事前面談にて確認してください。
| 相談の種類 | 相談内容及び留意点 |
| 既存の治験相談 | 申請電子データ提出に関連する事項としては、申請に必要な臨床データパッケージのうち、申請電子データ提出が必要な臨床試験等の特定について議論する相談。 臨床データパッケージ内の全ての臨床試験・解析に関する申請電子データを提出する場合は、事前面談等でその旨を審査部に連絡することでも差し支えない。 |
| 医薬品申請電子データ提出免除相談 | 治験相談において申請電子データの提出対象とされた個々の試験について、以下の内容等を議論する相談。
|
| 医薬品申請電子データ提出方法相談 | 申請電子データを提出予定の試験・解析に関して、以下の内容等を議論する相談。
|
| 医薬品申請電子データ提出確認相談 | 申請電子データ提出前に新医薬品承認審査予定事前面談の実施が想定されない状況での申請電子データの提出にあたり、必要に応じて事前に実施されたバリデーション結果に基づく、Errorに相当するバリデーションルール違反の説明と、修正が不可能な理由の確認を行う相談。 |
| 新医薬品承認審査予定事前面談 | 申請資料の提出内容及び提出予定時期について最終確認を行う相談。 |
Q1-6:(削除)
Q1-6-1:(削除)
Q1-6-2:(削除)
Q1-6-3:(削除)
Q1-7:承認申請時のゲートウェイシステムを用いた電子ファイルの提出が可能な期間を教えてください。また、提出予告時に指定した電子ファイルの提出予定日とは異なる日付でゲートウェイシステムを用いた電子ファイルの提出を行うことは可能でしょうか。
A: 電子ファイルを送信可能な期間は、ゲートウェイシステムで申請予告を作成した日から、申請予告時に指定した承認申請書提出予定日(申請予定日)までの期間です。申請予定日までであれば、提出予定日とは異なる日に電子ファイルを送信することが可能ですが、電子ファイルの送信日が提出予定日を過ぎる場合は、提出予告を修正してください。また、申請予定日を過ぎてから電子ファイルを送信することはできませんので、申請予定日までにファイルを送信してください。
なお、PMDA受付窓口にて承認申請を受け付ける際には、送信された全ての電子ファイルのウイルスチェックが完了し、FD申請データのバリデーションに問題がないことが確認されている必要があります。電子ファイルのウイルスチェック等を行う時間が必要となります。利用者が集中した場合は処理に時間を要しますので余裕をもったご対応をお願いします。
Q1-8:ゲートウェイシステムに「承認審査予定事前面談番号」を入力する箇所がありますが、何を参照して入力すればよいですか。
A: 新医薬品承認審査予定事前面談の受付番号を入力してください。新医薬品承認審査予定事前面談の受付番号は、PMDAの担当者より面談の日程を連絡する際等にお知らせします。
Q1-9:ゲートウェイシステムを利用することにより、照会事項及び照会事項回答のファイル作成方法に変更はありますか。
A: いずれのファイルの作成方法にも変更はありません。なお、照会事項回答のファイル容量については申請電子データシステムの操作マニュアルを参照してください。
Q1-10:(削除)
Q1-11:承認審査中の新有効成分含有医薬品に対し、申請中の効能とは異なる効能を追加する等の申請(以下「新規申請中の効能追加等申請」という。)を行う場合は、別途新有効成分含有医薬品として申請することとなります。新規申請中の効能追加等申請については、当該新医薬品が新有効成分含有医薬品として承認された後、一旦取り下げ、承認事項一部変更承認申請として再度申請することとなりますが、この場合、新規申請中の効能追加等申請時に提出した全ての申請電子データを再度提出する必要はありますか。
A: 必要です。ただし、新有効成分含有医薬品として承認を取得した際に提出していた申請電子データと重複する申請電子データについては、改めて提出する必要はありません。なお、この間に、新有効成分含有医薬品としての申請時には利用可能であった標準のバージョンの受付終了や、使用可能であったバリデーションルールのバージョンの受付終了があった場合、新有効成分含有医薬品としての申請時に提出し、効能追加等申請時に再度提出が必要になったデータに関しては、提出データ自体に変更がないのであれば、基本的には、特段の追加の対応は必要ありません。
Q1-12:(削除)
Q1-13:(削除)
Q1-14:(削除)
Q1-14-1:(削除)
Q1-14-2:(削除)
Q1-15:治験相談にて申請電子データの提出範囲に関する相談を行う際、どのような相談資料を提出すればよいですか。
A: 申請データパッケージに関する相談時と同様の資料に加え、申請電子データの提出範囲の妥当性について申請電子データ通知等に基づき説明してください。
Q1-16:バリデーションソフトウェア開発企業により、PMDAで用いるバリデーションソフトウェアと同等の結果を返すと公表されたバリデーションソフトウェアを用いてバリデーションを実施し、その結果に基づきReject又はErrorに相当するバリデーションルール違反に関してデータの修正又は必要な説明を行う場合、留意点はありますか。
A: 申請電子データの事前のバリデーションにおいて、バリデーションソフトウェア開発企業によりPMDAが用いるバリデーションソフトウェアと同等の結果を返すとされたバリデーションソフトウェアを用い、Rejectに相当するバリデーションルール違反について当該結果に基づいたデータの修正がなされていることが確認できた場合には、原則申請電子データを受領の上、FD申請データ及びeCTD等その他の申請資料に問題がなければ承認申請を受け付け、審査を開始することとしています。ただし、PMDAによるバリデーションにより、Rejectに相当するバリデーションルール違反が新たに検出された場合には、データの修正を求めます。また、申請者による事前のバリデーションで生じたErrorに相当するバリデーションルール違反については、違反が生じた理由及び修正が不可能な理由がデータガイドにおいて説明されている必要があります。
Q1-17:バリデーションソフトウェアの不具合によるものと考えられるバリデーションルール違反が検出されましたが、データを修正する必要はありますか。
A: バリデーションソフトウェアによりバリデーションルール違反が検出された場合であっても、データセットを確認した際、明らかに当該違反には該当しないと判断できる場合は、そのように判断した根拠をデータガイドで説明してください。なお、当該バリデーションルール違反が生じた原因がバリデーションソフトウェアの不具合である旨がバリデーションソフトウェア開発会社から公表されている場合は、この旨を判断した根拠として説明することで差し支えありません。
Q1-18:申請電子データを提出した後のPMDAにおけるバリデーションの実施方法、システムにおけるウイルスチェック、PMDAによるバリデーション結果の確認方法を教えてください。また、申請電子データの修正等が必要となった場合、どのように連絡されるのでしょうか。
A: PMDAでは、申請電子データの受信完了後、ウイルスチェック及び署名検証を実施し、これらに特段の問題がなければバリデーションを実施します。また、出力されたバリデーションレポート及びデータガイド等の内容に基づき、PMDA担当者がデータ受領可否について判断し、その結果をゲートウェイシステムに入力します。
署名検証終了後、バリデーションが実施され、データ受領可否の判断を行っている間はシステム上「バリデーション中」と表示され、データ受領可否判断の結果がシステムに入力された段階で「バリデーション完了」に遷移します。申請電子データが提出されてからデータ受領可否判断を終えるまでの期間は、通常5営業日程度を予定しています。
ウイルスチェック結果及びPMDAによるバリデーション結果は、それぞれウイルスチェック終了時点及びデータ受領可否判断の結果をPMDA担当者がシステムに入力した時点で確認できるようになります。具体的な確認方法については、申請電子データシステムの操作マニュアルを参照してください。
なお、バリデーションの結果、Rejectに相当するバリデーションルール違反が検出された場合、申請前であれば書面にて、申請後には照会事項として、対応が必要となる申請電子データ及びバリデーションルール等を連絡します。申請者による事前のバリデーションで生じたErrorに相当するバリデーション違反に関してデータガイドにおいて説明されていない場合には、申請後に照会事項により説明を求めます。
Q1-19:(削除)
Q1-20:ある品目の承認申請時に提出した申請電子データが審査の中でどのように活用されたのか、フィードバックを受けることはできますか。
A: 「新医薬品の承認審査の進捗状況の確認について」(平成22年12月27日付け薬機発第1227001号独立行政法人医薬品医療機器総合機構理事長通知)の別添の7において、審査終了後の適切な時期に申請者又はPMDAのうち、どちらか一方の要請があり両者が必要と認めた場合に、当該申請又は審査に関する問題点、今後の課題等について意見交換を試行として行うとされています。申請電子データに関するフィードバックもこの意見交換の中で実施いたしますので、必要に応じて担当審査部にご要望ください。
Q1-21:新医薬品の申請電子データの提出に係る相談の記録の写しを承認申請書添付資料として提出する必要はありますか。
A: はい。新医薬品の申請電子データの提出に係る相談を実施している場合は、医薬品申請電子データ提出確認相談(有料)では「医薬品申請電子データ提出確認相談結果要旨」、医薬品申請電子データ提出方法相談では「医薬品申請電子データ提出方法相談結果要旨」、医薬品申請電子データ提出免除相談では対面助言記録の写しを、CTDの第1部の「13.その他」の「(2)治験相談記録(写)」として提出してください。
Q1-22:データガイドにバリデーション結果を記載する際、Rule IDも記載する必要がありますか。
A: はい。バリデーション結果をデータガイドに記載する際は、PMDAが公表しているPMDA Study Data Validation RulesのRule IDをあわせて記載してください。
Q1-23:申請電子データ通知3(2)イに基づき、修正が不可能なErrorに相当するバリデーションルール違反に関する必要な説明をデータガイドに記入する際、留意点はありますか。
A: 修正が不可能なErrorに相当するバリデーションルール違反に関して、PMDAでは、データガイドの説明内容に基づいて審査時に実施する解析への影響の程度を検討していますので、そのような検討が可能となるようバリデーションル-ル違反の原因となったデータの実際の格納内容等を記載してください。また、企業側の解析に影響がないと説明する場合、その判断に至った理由を記載してください。なお、当該バリデーションルール違反の修正が不可能な理由についても漏れなく記入してください。
Q1-24:申請電子データに対してPMDAが実施するCDISC標準への適合性に関するバリデーションにおいて、どのような内容のバリデーションを実施していますか。
A: PMDAにおいては、個々のSDTM及びADaMデータセットのバリデーションに加え、SDTMデータセットとADaMデータセットの整合性に関するバリデーションも実施しています。また、define.xmlに関しては、define.xmlに対するXMLの構造等のバリデーションに加え、SDTM又はADaMデータセットの内容とそれぞれのデータセットに対するdefine.xmlの内容との間の整合性に関するバリデーションを実施しています。申請者による事前の適合性確認時にも同様のバリデーションを実施する必要があることに留意してください。
Q1-25:ゲートウェイシステムの「試験データ提出」画面で入力が必要なAnalysis Typeについて、入力する際の留意点を示してください。
A: 技術的ガイド4.2.1に記載されている「臨床薬理領域の電子データパッケージ説明書」を作成する際には、Analysis Typeで入力された情報を用いて臨床薬理領域のファイルを特定しているため、「臨床薬理領域の電子データパッケージ説明書」に適切な内容を含めるために、Analysis Typeを適切に入力する必要があります。
臨床薬理領域の解析に関する申請電子データを含まない試験又は解析については、全てのファイルでNon-CPを選択してください。
臨床薬理領域の解析に関する申請電子データを含む場合での申請電子データ提出対象となる資料の種類とAnalysis Typeとの関係を、申請電子データ通知における分類に基づいて示すと、以下の表1-25のとおりとなります。したがって、以下の考え方に基づき、各試験又は解析について、全てのファイルで同じAnalysis Typeを選択してください。ただし、1つの試験報告書に標準的な薬物動態解析及び母集団解析を含む場合は、母集団解析のファイルはPOPを選択し、他の全てのファイルはSTSを選択してください。また、母集団解析も実施した検証試験等で、母集団解析単独の報告書が存在しない場合には、当該試験について、母集団解析関係のファイルはPOPを選択し、他の全てのファイルはNon-CPを選択してください。
表1-25 臨床薬理領域の解析に関する申請電子データを含む場合の申請電子データ提出対象となる資料の種類とAnalysis Typeとの関係
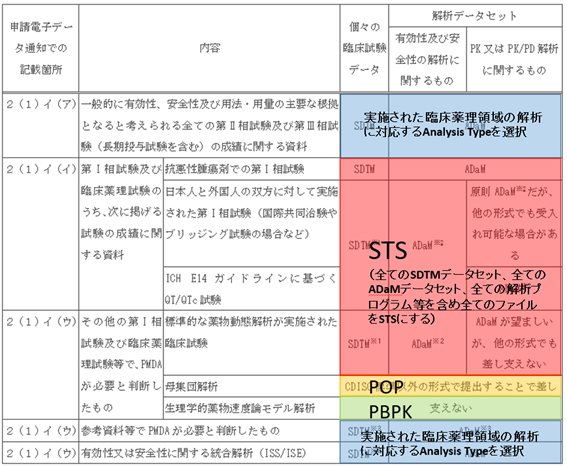
なお、STS、POP及びPBPKのいずれにも該当しないと考えられる臨床薬理領域の解析に関するファイルを提出する場合はOtherを選択してください。
Analysis TypeにおいてSTS、POP、PBPK又はOtherを選択したファイルについては、Descriptionを記載してください。ただし、STS又はOtherを選択したファイルのうち、CDISC標準に準拠した申請電子データ及び関連する文書(技術的ガイド4.1参照)については、Descriptionを「-」としても差し支えありません。
Q1-26:(削除)
Q1-27:(削除)
Q1-28:(削除)
Q1-29:申請電子データ通知Q&A問10「表 申請電子データ提出対象となる資料の種類と提出形式」で示されている内容に沿った形式のデータが提出可能な場合、医薬品申請電子データ提出免除相談の実施は不要と考えてよいですか。
A: 医薬品申請電子データ提出免除相談は、申請電子データ通知Q&A問2又は問15に基づき、試験データの一部又は全部を提出することが困難な場合に相談するものであり、申請電子データ通知Q&A問10に従ったデータが提出できるのであれば、医薬品申請電子データ提出免除相談は不要です。
Q1-30:申請電子データ通知4(2)イ(オ)において、SDTM以外の形式でまとめられている臨床試験データを承認申請時提出用にSDTM形式に変換する際に、データ取得時の設定により推奨される統制用語に合わせて変換できないデータがあるなど、部分的に困難な箇所がある場合には、対面助言を利用して事前にPMDAに相談すること、と記載されていますが、どの相談区分で相談すればよいでしょうか。
A: 医薬品申請電子データ提出方法相談で相談してください。
Q1-31:申請電子データ通知Q&A問18の第Ⅰ相試験、臨床薬理試験等の成績及び臨床薬理領域の解析に関する申請電子データの提出に関して、以下のように記載されています。それぞれどの相談区分で相談すればよいでしょうか。
- 申請電子データ通知Q&A問18(1)の2(1)イ(イ)に掲げる資料のうち、日本人と外国人の双方に対して実施された第Ⅰ相試験の有効性及び安全性の解析データセットについて、「必ずしも解析データセットの提出を要しない場合があるので、有効性及び安全性に関する解析データセットを提出することが困難な場合は、その提出要否を対面助言を活用し事前にPMDAに相談すること。」と記載されていること
- 申請電子データ通知Q&A問18(1)の2(1)イ(イ)に掲げる資料のうち、日本人と外国人の双方に対して実施された第Ⅰ相試験の薬物動態又は薬物動態/薬力学に関する解析データセットについて、「ADaM以外の形式で作成されているデータセットでも受入れ可能な場合があるので、薬物動態又は薬物動態/薬力学に関する解析データセットをADaM形式で提出することが困難な場合は、上記と同様に、事前にPMDAに相談すること。」と記載されていること
- 申請電子データ通知Q&A問18(2)の2(1)イ(ウ)の資料のうち、標準的な薬物動態解析が実施された臨床試験の有効性及び安全性の解析データセットについて、「必ずしも有効性及び安全性に関する解析データセットの提出を要しない場合があるので、当該解析データセットを提出することが困難な場合は、その提出要否を対面助言を活用し事前にPMDAに相談すること。」と記載されていること
- 申請電子データ通知Q&A問18(3)の複数の臨床試験のデータを統合したデータセットを用いて標準的な薬物動態解析を実施した場合について、「統合解析に用いた解析データセットに加えて、提出範囲となる個々の試験については、申請電子データの提出を求める。申請電子データの提出が困難な場合は、対面助言を活用し事前にPMDAに相談すること。」と記載されていること
A: 1. 既存の治験相談で相談してください。
2. 医薬品申請電子データ提出方法相談で相談してください。
3. 既存の治験相談で相談してください。
4. 医薬品申請電子データ提出方法相談で相談してください。
Q1-32:申請電子データ通知Q&A問19に関連して、「開発後期に実施された臨床試験のデータに基づく母集団解析等に関する申請電子データについては、申請後の提出も受入れ可能な場合がある。申請時に提出が困難な申請電子データがある場合は、具体的な提出時期を示した上で、当該申請電子データの提出が申請後になることについて、対面助言を活用し事前にPMDAに相談すること。」とありますが、どの相談区分で相談すればよいでしょうか。
A: 申請時に母集団解析等に関する申請電子データの提出が困難である場合、申請電子データに関わる特有の事情とあわせて医薬品申請電子データ提出方法相談で相談してください。
Q1-33:申請電子データ通知Q&A問16において、CDISC標準以外の形式で作成したデータをCDISC標準に変換して提出する場合の留意点について、「やむを得ない事情がある場合は、申請電子データ通知の2(1)イ(ウ)に該当する臨床試験であって、試験開始日(最初の被験者を組み入れた日)が平成26年6月20日より前の試験に限り、申請後の提出も受け入れ可能な場合があるため、対面助言を活用し事前にPMDAに相談すること。」と記載されていますが、どの相談区分で相談すればよいでしょうか。
A: 医薬品申請電子データ提出方法相談で相談してください。
Q1-34:申請電子データ提出時のデータ標準について、申請電子データ通知4(1)において「対象となる臨床試験のデータについては、CDISC標準に準拠した形式で提出すること。ただし、希少疾病用医薬品等について、令和2年4月1日より前に開始した試験については、その限りではない。」と記載されていますが、希少疾病用医薬品等について、統合解析のデータを提出する場合の取扱いを教えてください。
A: 統合解析のデータについても、解析報告書の作成日が令和2年4月1日より前であれば、CDISC標準以外の形式で提出することで差し支えありません。
Q1-35:承認申請時に、レジストリのデータ(以下、「レジストリデータ」)を臨床成績に関する資料として活用する場合の、電子データの提出内容について確認したい。
A: レジストリデータを臨床成績に関する資料として活用する場合であっても、基本的に、臨床試験の場合と同様、承認申請時に有効性、安全性及び用法・用量の主要な根拠となると考えられる資料の電子データをCDISC標準に準拠した形式で提出いただくことが重要であると考えます。
一方で、レジストリデータに関する電子データの提出範囲、提出内容及び提出形式については、レジストリデータの内容や活用目的により個別の判断を要すると考えられるため、対応する対面助言等において事前にPMDAに相談してください。
なお、データの提出形式に関しては、少なくともレジストリデータを臨床試験における外部対照として有効性等の評価に活用する場合で*、レジストリデータを利用した解析の内容が技術的ガイド4.1.1.3において解析データセットを提出することとしている解析に相当する場合には、当該解析に用いたデータセットは当該臨床試験の解析データセットの一部としてADaM形式で提出してください。解析方法やその他の技術的な理由からADaM形式で提出することが困難な場合には、医薬品申請電子データ提出方法相談で相談してください。
*参考:「承認申請等におけるレジストリの活用に関する基本的考え方」(薬生薬審発0323第1号・薬生機審発0323第1号、令和3年3月23日)
申請電子データとeCTDとの関連についての質問
Q2-1:ゲートウェイシステムからeCTDを提出した場合のeCTDカバーレターについて、電子媒体の種類、提出枚数の項の記載方法について確認させてください。
A: eCTD v3.2.2をゲートウェイシステムから提出する場合、カバーレターの「電子媒体の種類」は「ゲートウェイシステム」、「提出枚数」は半角ハイフン(「-」)としてください。
eCTD v4.0以降をゲートウェイシステムから提出する場合、カバーレターの提出は不要となりますが、XMLインスタンスのチェックサム値等必要な情報はゲートウェイシステムにて入力していただきます。
Q2-2:eCTDと申請電子データの提出日が異なることは許容されますか。また許容される場合、提出の順番に規定がありますか。
A: eCTDと申請電子データの提出日が異なることは差し支えありません。
eCTD v3.2.2を利用する場合、提出の順番に規定はありません。eCTD v4.0を利用し、①申請電子データのみを含めたeCTDと、②申請電子データ以外のCTD文書を含めたeCTDを分けて提出する場合は、②に対して①を先行して提出してください。詳細は、「電子化コモン・テクニカル・ドキュメント(eCTD)による承認申請について」(平成29年7月5日付け薬生薬審発0705第1号厚生労働省医薬・生活衛生局医薬品審査管理課長通知)の別紙1 「ICH電子化コモン・テクニカル・ドキュメント(eCTD) v4.0の国内実装について」を参照してください。
Q2-3:(削除)
Q2-4:申請電子データをeCTDのXMLメッセージから参照する場合以外において、申請電子データに変更(追加、置換、削除)がある場合、その都度eCTDを改訂する必要があるのでしょうか。
A: 申請電子データの変更の有無に関わらず、PMDAが必要と判断した場合は部会前に限らずeCTD改訂版を提出していただくことは従来どおりです。申請電子データに変更がある場合であっても、変更の内容や申請・審査業務効率の観点から、都度の改訂を求めず複数の変更を一回の改訂に纏めることで差し支えない場合もあります。eCTD改訂版を提出するタイミングについては担当の審査チームにご相談ください。
Q2-5:(削除)
Q2-6:技術的ガイド5.5②「eCTD改訂時に提出する申請電子データ」に「既提出の申請電子データとの差分のみを提出する。」とありますが、例えば24週時点までの臨床試験成績の申請電子データを申請時に提出し、同一臨床試験の52週時点までの申請電子データを審査中に提出する場合に、差分のみを提出するというのは困難な場合があります。このようなケースにおいて、どのように申請電子データの差分を提出すべきでしょうか。
A: eCTD改訂時には、既提出の申請電子データとの差分のみを提出することとしていますが、ここでいう「差分」とは、試験又はファイル単位での差分を指します。したがって、審査期間中に変数又はレコードが追加されたデータセットは、既提出の変数及びレコードも含め最新版のデータセットとして提出してください。
Q2-7:(削除)
Q2-8:(削除)
Q2-9:(削除)
Q2-9-1:(削除)
ゲートウェイシステムについての質問
Q3-1:(削除)
Q3-2:(削除)
Q3-3:操作中にゲートウェイシステムのエラーが発生した場合、どこに連絡をしたら良いでしょうか。
A: ゲートウェイシステムのトップページに掲載している問い合わせ票内に問合せ先の窓口メールアドレスが記載されていますのでご確認ください。ゲートウェイシステムにログイン後であれば、操作マニュアルに従いシステムのお問合せ機能から問い合わせすることも可能です。なお、ヘルプデスクの受付時間により、返信に時間を要する可能性がありますのでご了承ください。
Q3-4:ゲートウェイ申請において、ゲートウェイシステムから提出可能な電子ファイルは何でしょうか。
A: ゲートウェイシステムから提出可能な電子ファイルの例を以下の表3-4に示します。
| FD申請データのうち以下2種類: 医薬品製造販売承認申請書* 医薬品製造販売承認事項一部変更承認申請書* |
| eCTD |
| 申請電子データ** |
| 申請資料作成関与委員リスト |
| 競合品目・競合企業リスト |
| 競合品目関与委員リスト |
| 添加物換算係数CSV |
| 照会事項に対する回答 |
| 医薬品第一部会及び第二部会のPMDAへの提出資料 |
* 電子ファイルとは別に書面での提出も必要となります(審査中の差換え時も同様)。
**技術的ガイド別紙1参照
Q3-4-1:Q3-4に関連して、表3-4の各電子ファイルについて、書面提出の要否や提出時期の考え方を示してください。
A: 通知等により書面提出が必要とされている文書については、従来どおり書面の提出が必要となります。
また、通知等により申請に際し提出することとされていた文書の提出時期について、申請前にゲートウェイシステムを利用して当該電子ファイルを提出することは差し支えありません。
Q3-5:(削除)
Q3-6:(削除)
Q3-7:試験データを格納する場合のフォルダ構造に関して、技術的ガイド3.5において、[study id / iss / ise]とありますが、具体的には何でしょうか。
A: [study id / iss / ise]は、申請者が各臨床試験に対して設定したIDを指します。当該IDは各試験を一意に識別できる試験番号又は解析の種別であれば申請者が自由に決めることができますが、eCTDを提出する場合であり、かつ申請電子データをeCTDのXMLメッセージから参照する場合以外は、eCTDのM5の総括報告書のフォルダ名と同一の名称にしてください。
Q3-7-1:試験データを格納する場合のフォルダ構造及び格納する情報の留意点を教えてください。
A: 基本的には、eCTDのM5の総括報告書を格納するフォルダと試験データを格納するフォルダについて、それぞれに格納する情報を1対1で対応させることが必要です。
フォルダ構造及びそれぞれに格納する情報を1対1にする上で特に注意が必要な場合として、例えば、以下のような対応が考えられます。
- 臨床試験において、1つの総括報告書が作成されるが、当該臨床試験に関する申請電子データについては臨床試験データセット(解析データセットが付随する場合もある)の複数組を提出する場合(例:評価時点ごとに臨床試験データセットを作成する場合)、申請電子データについては、各組に対応するフォルダを作成した上で、それぞれの申請電子データを格納してください。総括報告書を格納するフォルダも各申請電子データを格納するフォルダに対応するよう作成した上で、それぞれに同一の総括報告書を格納してください。または、一方のフォルダに総括報告書を格納し、他方のフォルダには総括報告書を格納したフォルダを参照する旨を記した文書(PDFファイル)を格納してください。
- 同一申請内において、試験IDが異なる複数の報告書間で対応する申請電子データが共通している場合、報告書については、各報告書に対応するフォルダを作成した上で、それぞれの報告書を格納してください。申請電子データを格納するフォルダも各報告書を格納するフォルダに対応するよう作成した上で、同一の申請電子データをそれぞれのフォルダに格納してください。または、一方のフォルダに申請電子データを格納し、他方のフォルダ下の「misc」フォルダには、申請電子データを格納したフォルダを参照する旨を記した文書(PDFファイル)のみを格納してください。
Q3-8:各種電子ファイルを送信する際に、当該電子ファイルを暗号化する必要はあるでしょうか。
A: 電子ファイルの送信経路は、システムにより自動的に暗号化されますので、申請者側で電子ファイルを暗号化する必要はありません。
Q3-9:(削除)
Q3-10:申請電子データに誤りがあった場合、差換え提出は可能でしょうか。また、可能である場合、誤りがあった部分のみを差し換えることでよいでしょうか。
A: 申請電子データを申請者の都合により差し換える場合は、PMDAがロック解除の操作をすることで、申請者による差換えが可能となります。このような場合は、審査部の担当者にご連絡ください。
なお、申請電子データの差換え提出範囲は、提出のタイミングやバリデーションの状態によって変わりますので、以下の表3-10を参照して提出してください。
| 差換え提出時期 | |||
| 承認申請受付前 | 承認申請受付後 | ||
| 提出した申請電子データの受領可否 | 受領可の場合 | 申請電子データ全体 | 誤りがあった電子ファイルのみ(改訂として提出) |
| 受領不可の場合 | 申請電子データ全体 | 申請電子データ全体 | |
Q3-11:ゲートウェイシステムの申請予告の入力画面に、GLP及びGCP関連書類の提出の有無という項目が設けられています。これらの項目に関する資料は、何を想定すればよいでしょうか。
A: 申請予告作成画面は、新医薬品承認申請前に審査業務部宛に送付いただいているFAXの記載内容に沿って入力していただくことを想定していますので、承認申請時における調査関連資料の提出有無を入力してください。なお、これまでにご案内しているように、ゲートウェイシステムを用いてGLP及びGCP関連調査資料の電子ファイルを提出いただくことは現在想定しておりません。
Q3-12:ゲートウェイ提出可能な電子ファイルの容量の上限はありますか。
A: ファイルサイズの上限については申請電子データシステムの操作マニュアルを参照してください。
Q3-13:提出可能な電子ファイルサイズの上限を超えた場合の対応方法を教えてください。
A: 1ファイルのサイズが上限値を超える場合の対処方法について、以下に例示します。
- ファイル圧縮する:
(例)「その他」ファイルをzip化してサイズを縮小し提出する。その際、暗号化はしない。
- 他の提出経路を用いる:
(例)照会回答の添付ファイルが上限値を超える場合に、「その他」ファイルとして提出する。
- ファイルをページで区切って作成する:
(例)eCTDのPDFファイルが上限値を超える場合、文書の途中でファイルを分けて上限値以下のファイルを複数作成する。
申請電子データに含まれるPDFファイル、eCTD、添加物換算係数CSV、「その他」ファイル、及び照会事項回答として提出するファイルにおいて、上記の方法では対応が困難な場合は、ファイルを分割してそれぞれ送信してください。その際、ファイル復元(結合)方法を併せて遅滞なくPMDAに連絡してください。
Q3-14:(削除)
Q3-15:ゲートウェイシステムがメンテナンスにより停止されることはあるのでしょうか。また、システムダウンしている旨は、どのように周知されるのでしょうか。
A: システムの健全性を保つため、適宜メンテナンスを実施することとしています。これらは事前に計画されるものですので、4週間前を目処にゲートウェイシステムのトップページにてお知らせいたします。
なお、トップページを含むシステムダウンが発生し、上記連絡手段が使用できない場合はPMDAのWEBサイト(https://www.pmda.go.jp/)にて情報提供を行います。
Q3-16:(削除)
Q3-17:(削除)
Q3-18:ゲートウェイ申請において、承認申請書のFD申請データを、ゲートウェイシステムを用いて提出し、ウイルスチェック及びバリデーションは完了した後に、承認申請書のPMDA窓口提出時にFD申請データを含めた承認申請書の修正が求められる場合はありますか。
A: あります。その場合は、申請電子データシステムの操作マニュアルに従い修正したFD申請データを、ゲートウェイシステムで提出してください。その後、同内容の承認申請書を窓口提出してください。
Q3-19:(削除)
Q3-20:申請後に追加で申請電子データのデータセットを提出する場合の提出方法及び留意点を示してください
A: 申請後の申請電子データのデータセットの追加提出の方法及び手段は、以下が考えられます。
<追加提出の方法>
- 新たなデータセットを追加提出する
- 既提出のデータセット中の情報を追加(又は情報を更新)しデータセットを置換する
<追加提出の手段>
- CTD/eCTDを改訂する
申請後に申請電子データを追加提出する際は、いずれの方法・手段の場合でも、先の提出時と同様に技術的ガイド3.5に提示しているフォルダ構造にデータセットを格納し、ゲートウェイシステムの操作マニュアルに記載のとおりの手順で、ゲートウェイシステムを通じて、追加又は変更のあったデータセットを提出することになります。また、変更があった特定のSDTMドメインやADaMデータセットのみではなく、データセットの追加・変更に伴って追加・変更が発生した文書等(データセットに付随して提出すべき文書や解析用プログラム等)があれば、それらも併せてご提出ください。
なお、PMDA側ではデータ受領のタイミング毎に上位フォルダを分けて管理しています。したがって、追加提出データを既提出フォルダと同じ名称のフォルダに格納して提出したとしても、既提出の申請電子データは追加提出データで上書きされることはなく、バージョン管理されることになります。
申請後に継続している臨床試験の申請電子データを審査期間中に追加提出してもらうことを一律には求めませんが、審査をするうえで追加提出が望ましい場合もありますので、追加提出の要否や提出時点については審査チームと相談してください。
Q3-21:(削除)
Q3-22:(削除)
Q3-23:複数の試験の申請電子データを1回の操作で1度に提出可能でしょうか。
A: はい、1回の操作で1度に複数の試験を提出可能です。
Q3-24:ゲートウェイ申請において、承認申請にあたり、やむを得ない事情により、ゲートウェイシステムで提出する予定であった電子ファイルを、PMDA窓口に提出する予定です。その際の留意点を教えてください。
A: ゲートウェイシステムを用いて電子ファイルの送付を試みたものの、やむを得ない事情によりPMDA窓口に提出する場合は、以下の点に留意する必要があります。
(事前提出時)
- ゲートウェイシステムを用いた申請予告は行うようにしてください。
- 提出予告情報の「提出方法」欄は、「ゲートウェイ」を選択してください。
- 申請予定日2勤務日前までに、ヘルプデスクに、担当審査部と品目名、窓口提出を希望する旨、及び記録媒体の窓口提出希望日(遅くとも申請予定日の1勤務日前まで)を連絡し、事前にPMDAの了承を得てください。連絡の際、記録媒体が2枚以上にわたる場合はその旨も連絡してください。なお、窓口提出の希望を連絡した後も、窓口への提出日まではなるべくゲートウェイシステムでの提出を試みるようにしてください。また、申請予定日の1勤務日前に窓口に提出する場合は、受付処理のために、なるべく午前中に提出するようにしてください。
- 窓口提出の際には、ゲートウェイシステムで提出する予定であった申請資料を格納した記録媒体及び申請予告受付票(紙)を準備し、審査業務部業務第一課に持参又は郵送して提出してください。記録媒体の表面には、ゲートウェイ受付番号、申請者名、販売名及び申請予定日を、申請予告受付票(紙)には赤字で「GW登録」と記載してください。取違えのリスクを防ぐため、受付票がない場合等、品目が特定できない場合は記録媒体を受領できないことに留意してください。なお、記録媒体の窓口提出にあたっては、審査業務部業務第一課に事前に連絡する必要はありません。
- FD申請データ、eCTD、申請電子データ及びその他資料について、複数種類を提出する場合は、それぞれ記録媒体を分けて提出してください。なお、ゲートウェイシステムにより送信可能であった種類の電子ファイルについては窓口に提出せず、送信できなかった種類の電子ファイルのみを窓口に提出してください。また、申請電子データ以外の電子ファイルについて、複数種類を窓口に提出する場合は、一度に纏めて提出してください。
- 複数品目を同時申請する場合は、FD申請データのファイル名と品目名が一意に紐付けられる情報(対応表)をご提示ください。
- eCTDをDVDに保存して提出する場合、複数枚に分割して保存しても差し支えありません。ただし、多層ディスクを利用する等媒体は可能な限り1枚に収めてください。なお、eCTD受付番号を再度取得する必要はなく、申請予告時に取得したものを用いてください。
- 申請電子データは、複数枚の記録媒体に分割して提出された場合にPMDA側で本来のフォルダ構造を再現すること、及び再現できたかを確認することが困難であることから、BD(多層ディスクを含む。)を利用する等し、原則として1枚に収めてください。なお、二層式のDVD-Rや多層式のBD-R/RE 等を用いた場合であっても1枚に収まらない場合は、個別に相談してください。
- eCTDのXMLメッセージから参照する場合を除き、申請電子データ提出に際して、臨床試験データ(申請電子データ)の提出内容を示すタブ区切り形式(TSVファイル)の提出が必須となるため、臨床試験データ提出内容を示すTSVファイルを作成し、「m5」フォルダと同パスに配置して提出してください。なお、TSVファイルの作成方法については、PMDAのWEBサイト(https://www.pmda.go.jp/review-services/drug-reviews/about-reviews/p-drugs/0028.html)に別途掲載するので参照するとともに、技術的ガイド3.5に定めるデータセットのファイル名に係る規定に従い任意のファイル名を付与してください。
- 提出された記録媒体は、原則としてPMDAにて廃棄します。返却を希望する場合は、申請予告受付票にその旨を記載してください。
(申請予定日)
- 事前に、PMDAが承認申請に併せて提出すべき全ての電子ファイルについて、ウイルスチェックにより当該ファイルに感染等の問題がないことを確認し、FD申請データのバリデーションに問題がないことが確認された場合、その確認が完了した旨の連絡をいたします。当該連絡後に、書面で提出すべき申請書類を審査業務部業務第一課に持参又は郵送して提出してください。
Q3-24-1:Q3-24に関連して、申請電子データ通知2(1)ア「保険衛生上の危害の発生及び拡大の防止のために緊急に使用されることが必要な医薬品」であって、申請時にゲートウェイシステムを使用せずに申請電子データを任意提出する場合の、提出方法を教えてください。
A: 申請資料を格納した記録媒体及び申請予告受付票(紙)を準備し、審査業務部業務第一課に持参又は郵送にて提出してください。記録媒体の窓口提出にあたっては、審査業務部業務第一課に事前に連絡する必要はありません。記録媒体の表面には、ゲートウェイ受付番号、申請者名、販売名及び申請予定日を記載してください。申請予告受付票には、赤字で「GW登録」と記載してください。取違えリスク防止のため、受付票がない等、品目が特定できない場合は記録媒体を受領できませんので、ご留意ください。なお、提出された記録媒体は、原則としてPMDAにて廃棄します。返却を希望する場合は、申請予告受付票にその旨を記載してください。
Q3-25:申請予告時に入力した承認申請書提出予定年月日よりも前に申請する場合、申請予告を修正する必要はありますか。
A: 承認申請書の提出予定日時の変更を希望される場合は、審査業務部業務第一課に連絡し日程調整の上、申請予告の登録内容を修正してください。なお、提出予告されたいずれかの電子ファイルがゲートウェイ提出されている場合は、予定日時の修正にPMDA側の対応が必要となりますので、上記連絡の際にその旨お伝えください。
Q3-26:(削除)
Q3-27:(削除)
Q3-28:ゲートウェイ申請において、FD申請ソフトでFD申請データを提出用に出力する場合、以下の二つの出力の形式が選択可能ですが、ゲートウェイシステムで提出する場合の出力方法をどうすればよいでしょうか。
- CD-R焼込用ファイル出力
- オンライン申請用出力
A: ゲートウェイシステムで提出するFD申請データには、「CD-R焼込用ファイル出力」で出力したデータをご使用ください。
Q3-29:PMDA窓口で電子ファイルを提出する場合、TSVファイルの不備により、申請が受け付けられないケースはありますか。
A: はい、TSVファイルの不備により申請が受け付けられないケースはありえます。申請電子データが窓口提出された場合、PMDAでゲートウェイシステムを用い、①TSVファイルの取込み、②TSVファイル(同時に提出される申請電子データのフォルダ構造との整合性を含む。)の検証及び保存を行い、問題なければ③申請電子データの登録の操作を行います。ご質問の「TSVファイルの不備」がTSVファイルの取込み時又は検証機能により検出されるものである場合、これらの操作時にエラーが生じることから申請電子データの登録操作が完了せず、ウイルスチェックが完了した状態にならないため、申請を受け付けることができません。
Q3-30:申請電子データのバリデーション結果の「OK」と「受領可」の違いは何ですか。
A: バリデーションの結果、バリデーションルール違反が検出されなかった場合に「OK」と表示されます。一方、バリデーションの結果、バリデーションルール違反が検出されたものの、事前の説明内容等を踏まえ当該申請電子データを受領して差し支えないと判断した場合に「受領可」と表示されます。
Q3-31:ゲートウェイ提出した全ての電子ファイルについて、ゲートウェイシステム上でウイルスチェックや署名検証が完了しており、かつPMDA窓口での提出物に不備がなければ、バリデーション結果が得られていない場合であっても、承認申請は受け付けられますか。
A: FD申請データについては、承認申請の受付時までにバリデーションが完了し、結果が「OK」である必要があります。一方、eCTD及び申請電子データのバリデーション結果は承認申請受付の可否の判断には利用しません。
Q3-32:(削除)
Q3-33:申請電子データ通知Q&A問7に関連して、新医薬品の承認申請や再審査申請に先立って申請電子データを提出する場合、提出する申請電子データの形式や提出方法について、教えてください。
A: 相談、HIV感染症治療薬等の承認申請前、承認条件解除時等に任意で申請電子データを提出する場合、以下の点に留意してください。
(データセット)
- 申請電子データの提出対象となる試験・解析については、事前に審査部と相談してください。
- 申請電子データ提出に際して、臨床試験データの提出内容を示すタブ区切り形式(TSV)ファイルの提出が必須となるため、臨床試験データの提出内容を示すTSVファイルを作成し、「m5」フォルダと同パスに配置して提出してください。なお、TSVファイルの作成方法については、PMDAのWEBサイト(https://www.pmda.go.jp/review-services/drug-reviews/about-reviews/p-drugs/0028.html)に別途掲載するので参照するとともに、技術的ガイド3.5に定めるデータセットのファイル名に係る規定に従い任意のファイル名を付与してください。
- フォルダ構造は技術的ガイド3.5で規定する方法に準じて作成してください。総括報告書を格納するフォルダ名は試験データを格納するフォルダ名[study id / iss / ise]と同一にし、総括報告書を格納するフォルダと試験データを格納するフォルダについて、それぞれに格納する情報を1対1で対応させてください。
- 提出する申請電子データは、原則としてフォルダ構造を維持した1つのzipファイルにして提出してください。ただし、zipファイルのファイルサイズが申請電子データシステムの操作マニュアルに示す上限(ゲートウェイシステムで提出する場合)、又は記録媒体の上限(記録媒体で提出する場合)を超える場合は、試験ごと等に分割し、複数のzipファイルで提出してください。なお、データセットの分割が必要となる場合は、ヘルプデスクに連絡してください。
- 他の審査・相談資料と同じ提出先に提出してください。
- 相談時は、原則として相談申込書の提出日から相談資料搬入予定日までの期間に、ゲートウェイシステム又は記録媒体により申請電子データを審査マネジメント部マネジメント課に提出してください。相談資料搬入日と異なる日に申請電子データを提出する場合、事前に提出予定日を審査マネジメント部審査マネジメント課(shinyaku-uketsuke[at]pmda.go.jp([at]を半角のアットマーク@に置き換えてください。))宛てに電子メール等で連絡してください。なお、相談資料搬入日に申請電子データを提出する場合は、事前連絡は不要です。
- ゲートウェイシステムを利用して申請電子データを提出する場合、ゲートウェイシステムの提出名称(又は通信欄)に、申請電子データを提出する旨を記載してください。
- 記録媒体で申請電子データを提出する場合、BD(多層ディスクを含む。)を利用する等し、原則として1枚に収めてください。また、記録媒体の表面又は送付状に、申請電子データを提出する旨を記載し、記録媒体の表面には、相談区分、相談者名、受付番号、販売名(一般名)を記載してください。 記録媒体は、原則としてPMDAにて廃棄します。返却を希望する場合は申込書等にその旨を記載してください。
Q3-34:(削除)
CDISC標準に準拠した申請電子データについての質問
Q4-1:「申請電子データ提出に際して利用可能な規格一覧」における「受付開始時期」、「受付終了時期」とは何の日付が基準となりますか。
A: 申請者が申請書のFD申請データに記載する「提出年月日」が基準となります。
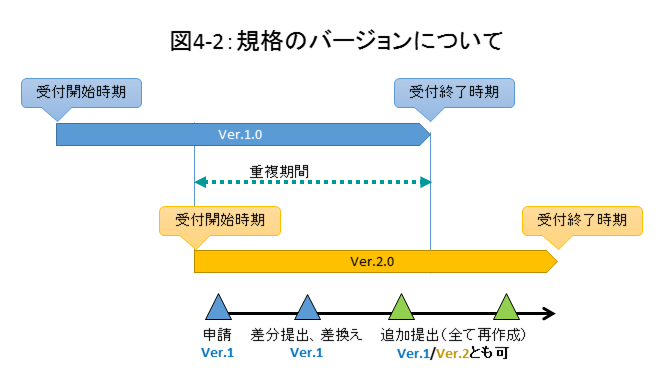
Q4-2:審査中の差換え、追加提出の際の規格のバージョンは申請時と同じにすべきでしょうか。また、その際に申請時に提出したバージョンの受付が終了していた場合はどうすべきでしょうか。
A: 申請時に受け付けたバージョンでの提出が基本となります。既に提出している申請電子データの差分を提出する場合は、既提出のバージョンに揃えてください。
また、バージョンの重複期間中に申請(初回の提出)をした以下のようなケースでは、申請時に利用したバリデーションルールに含まれていた新しい規格のバージョンで提出することも可能です。
- 追加で新規の申請電子データを提出する場合
- 既提出の申請電子データについて差分も含め全てを新しいバージョンで作成しなおして提出するような場合
詳細は図4-2を参照してください。
Q4-3:PMDAで実施するCDISC標準に準拠したデータに対するバリデーションの対象範囲を示してください。
A: PMDAでは、SDTMデータセット及びADaMデータセット、並びにデータセットの定義書、統制用語及び外部辞書の内容を対象として、CDISC標準に準拠したデータに対するバリデーションを実施します。日本語を含むデータセットが併せて提出される場合には、日本語を含むデータセットはバリデーションの対象とはせず、対応する英数字のデータセットのみをバリデーションの対象とします。
Q4-4:統合解析(ISS/ISE)に関する申請電子データの提出すべき内容を示してください。
A: ISS/ISEに関する申請電子データの提出に際しては、統合解析に用いられたデータセットに加えて、データセットの定義書及び解析用プログラム(又はプログラム仕様書)を提出する必要があります。また、データガイドも原則として提出する必要がありますが、ISS/ISEに関する申請電子データの提出形式によっては提出しないことでも受け入れ可能な場合があります。提出すべきファイルの種類等の詳細に関して確認したい場合は医薬品申請電子データ提出方法相談において相談してください。
Q4-5:(削除)
Q4-6:治験実施計画書等が日本語で作成されている場合、SDTMのTrial Design Modelのドメインには、臨床試験の計画に関する情報を英語に翻訳した上で格納する必要があるでしょうか。また、SDTMのTSドメインについて、PMDAが最低限提出を求めるパラメータ及びコードを示してください。
A: Trial Design Modelに格納されるデータについては、日本語を英語に変換した場合に損なわれる情報は大きくないと考えられます。よって、治験実施計画書等が日本語で作成されている場合にも、各情報を英語に変換した上で格納する必要があります。
SDTMの作成にあたり、SDTM IG v3.1.3以降に基づく場合は、TSドメインにRequired又はConditionally Requiredに分類されるパラメータを含めてください。SDTM IG v3.1.2に基づく場合は、SDTM IGの7.6.2-4項を参考に格納可能なパラメータを含めてください。統制用語、及びISOコードを用いるパラメータには、適切なコード値を格納してください。Registry Identifierには、CLINICALTRIALS.GOV、EUDRAC、又は JAPIC、jRCT等の登録番号を格納してください。UNII、NDF-RT、MED-RT、DUNS、SNOMED CTを用いるパラメータは、申請者が使用可能なコードの値のみを格納することで差し支えありません。統制用語、ISOコード以外のコードを用いるパラメータ及びコードの関係を表4-6に示します。
| TSPARMCD | TSPARAM | コード | WEBサイト |
|---|---|---|---|
| CURTRT | Current Therapy or Treatment | UNII | FDA's Global Substance Registration System https://precision.fda.gov/uniisearch |
| TRT | Investigational Therapy or Treatment | UNII | FDA's Global Substance Registration System https://precision.fda.gov/uniisearch |
| PCLAS | Pharmacological Class of Invest. Therapy | NDF-RT/MED-RT | NCI Term Browser https://nciterms.nci.nih.gov/ncitbrowser/ pages/multiple_search.jsf |
| REGID | Registry Identifier | CLINICALTRIALS. GOV | ClinicalTrials.gov https://clinicaltrials.gov/ |
| EUDRAC | EU Clinical Trials Register https://www.clinicaltrialsregister.eu/ctr-search/search |
||
| JAPIC | JAPIC Clinical Trials Information https://www.clinicaltrials.jp/cti-user/common/Top.jsp |
||
| jRCT | 臨床研究等提出・公開システム https://jrct.niph.go.jp/ |
||
| SPONSOR | Clinical Study Sponsor | DUNS | 東京商工リサーチ https://duns-number-jp.tsr-net.co.jp/search/jpn/login.asp |
Q4-7:申請電子データ通知4(2)エに「薬剤についてはWHODrug Globalを使用」とありますが、WHODrug Globalの利用を必須とすることの背景を説明してください。また、SDTMのCMドメインへのWHODrug Globalデータの格納方法について例示してください。
A: 臨床試験データの国際的標準化の推進と、将来的に予定している品目横断的解析の実施のため、申請時に提出する申請電子データにおいては共通してWHODrug Globalを利用することを求めます。なお、WHODrug Globalの内容を網羅した上で、WHODrug Globalに存在しないものについては独自のコード等を使用することは可能ですが、その場合、一部で独自のコード等を使用した旨をデータガイドで説明してください。
SDTMのCMドメインへのWHODrug Globalの各項目の格納方法の例を表4-7に示します。なお、WHODrug GlobalのATCコードも可能な限り格納してください。また、併用薬剤の使用目的を収集していない等の理由でWHODrug GlobalのATCコードを一意に特定できない場合、単一のATCコードのみを格納するのではなく、該当する全てのATCコードを、Supplemental Qualifier special-purpose datasetを用いて格納してください。
| Variable Name | Variable Label | WHODrug Global |
|---|---|---|
| CMDECOD | Standardized Medication Name | Generic name |
| CMCLAS | Medication Class | ATC text |
| CMCLASCD | Medication Class Code | ATC code |
Q4-8:申請電子データ通知4(2)エに「慣例的に使用されている単位によりデータが取得されている場合には、SI単位への変換が可能なものは、SDTMデータセット内に標準単位による値としてSI単位に変換後の値を別途格納して提出すること」とありますが、SI単位への変換対象となる変数の範囲を示してください。また、SI単位以外の単位のデータが取得された場合における、取得された単位のデータとSI単位データ(変換後)のそれぞれのSDTMへの格納方法を示してください。
A: SDTMのデータセットのうち、Findingsクラスのドメインに格納される検査項目に係る全ての変数及びパラメータについて、SI単位が設定されているものについてはSI単位での格納を求めます。ただし、慣例単位としてmmHgを使用してデータが取得されている検査項目(血圧等)については、SI単位への変換をせず、mmHgによるデータのみをSDTMに格納することで差し支えありません。
取得された単位のデータとSI単位のデータを併せてSDTMに格納する場合は、SI単位のデータは--STRESC(必要に応じて--STRESN)に格納することとし、取得された単位のデータは--ORRESに格納してください。また、データガイド又はデータセットの定義書において取得された単位のデータ及びSI単位のデータそれぞれの格納方法と単位間の変換式を説明してください。
また、例えば多施設共同試験や国際共同試験にて施設や地域毎に異なる測定単位でデータを取得する等、同一パラメータ(検査項目)内で複数の単位でデータが取得される場合があります。その際は、必要に応じてSI単位以外の単位で統一したデータをSUPP--に格納することは可能です。
<慣例単位データ及びSI単位データのSDTMへの格納方法の例>
国内の慣例単位の値と国外の慣例単位の値が存在する場合
--ORRES及びSUPP--に国内の慣例単位の値及び国外の慣例単位の値をそれぞれ格納する。SI単位の値は-STRESC(必要に応じて--STRESN)に格納する。
Q4-9:解析帳票において日本語で表示させたい変数については、データセットに日本語を格納することとしたいです。この場合、申請電子データ提出にあたっては、英数字のみからなるデータセットを別途用意する必要があるでしょうか。
A: 申請電子データの提出にあたっては、英数字のみからなるデータセットを提出することを基本としてください。言語間で情報を損なうことなく適切な翻訳(解析帳票上の日本語から英語への変換)が可能な変数であれば、申請時に提出するデータセットには英語を格納し、英数字のみのデータセットを提出することで構いません。このとき、解析帳票作成時に利用したデータセットと英数字のみのデータセットとの間のトレーサビリティを説明する観点、あるいは、審査において参考となる資料を提出するとの観点から、英数字のみのデータセットに加えて、翻訳された英数字による値の代わりに解析帳票作成時に利用した翻訳前の日本語が格納されたデータセットを併せて提出することは可能です。一方、日本語から英語に変換する際に一定の情報が損なわれる恐れのある場合は、技術的ガイド4.1.5に従い申請電子データを提出してください。なお、データセット作成時に言語間の翻訳がなされた時は、その旨データガイドで説明してください。
Q4-10:データセットに英語でも日本語でもない他の言語が入る可能性のある場合はどのように対応したらよいでしょうか。
A : 基本的には内容が全て英語に変換されたデータセットを提出することを求めます。特定の変数の英語への翻訳の必要性については、該当変数の重要度にも依存する可能性がありますので、個々のケースについては必要に応じて医薬品申請電子データ提出方法相談を利用してください。なお、言語間の変換がなされた場合はその旨をデータガイドで説明してください。
Q4-11:言語間の翻訳(例:日本語→英語)が発生した場合、その翻訳の適切性を証明する必要はありますか。
A : 翻訳に関する証明書等の提出は特段求めませんが、翻訳の適切性は申請者によって確保してください。
Q4-12:(削除)
Q4-13:「解析アルゴリズムの分かる仕様書等」に記載すべき内容の例を示してください。
A: 「解析アルゴリズムの分かる仕様書等」には、解析対象となるデータセット及び変数と解析手法の詳細を具体的に記載してください。
Q4-13-1:解析アルゴリズムの分かる仕様書を英語で提出することは可能ですか。
A: 可能です。
Q4-13-2:明示的なプログラムの作成を必要としない解析ソフトウェアを用いた場合は、操作ログの提出をもってプログラムの代わりとしてもよいでしょうか、あるいは、「解析アルゴリズムの分かる仕様書等の提出」が別途必要となるのでしょうか。
A: 操作ログに基づき解析アルゴリズムが分かる場合は、操作ログの提出をもってプログラムの代わりとすることが可能です。一方、操作ログのみでは解析アルゴリズムが分からない可能性がある場合、「プログラム自体の提出が困難な場合」となると判断し、「解析アルゴリズムの分かる仕様書等の提出」が別途必要となります。
Q4-14:(削除)
Q4-15:統合解析(ISS/ISE)に関する申請電子データを提出する場合のデータの格納先のフォルダを示してください。
A: 基本的には、統合解析の対象となる複数の試験のデータを1つに統合し、各解析について1つの解析データセットが提出されることを想定していますが、各試験のデータセットをプログラムにより統合して解析を実施している場合には、各試験のデータセット及び統合の過程を含むプログラムを提出することも可能です。なお、統合解析用のデータセットは、統合されているか試験毎かにかかわらず、「iss / ise」フォルダに格納してください。
Q4-16:解析用プログラムの提出が困難である場合、解析アルゴリズムを示した仕様書を提出しようと思いますが、いずれのフォルダに提出すべきでしょうか。
A: 解析の仕様書は「programs」フォルダへ格納してください。また、その旨をデータガイドで説明してください。
Q4-17:(削除)
Q4-18:(削除)
Q4-19:技術的ガイド4.1.1.4に「データセット作成時に用いられた文字セット又は符号化方式をデータガイドに含めること」とありますが、申請時に提示すべき文字セット及び符号化方式の情報の具体例を示してください。
A: データセットの作成者が意図した文字を特定するため、使用された文字セット又は符号化方式の情報が必要となります。
データガイドに記載されるべき文字セット情報の例
【文字セット】JISX0208 【符号化方式】Shift-JIS
【文字セット】JISX0208 【符号化方式】EUC-JP
【文字セット】UNICODE 【符号化方式】UTF-8
Q4-19-1:Q4-19に関連して、使用された符号化方式の情報はどのようにして得ればよいでしょうか。
A: 原則として、データセットのプロパティから符号化方式を得てください。なお、対象となる文字セットを一般とは異なる符号化方式でエンコーディングしている場合には、追加でより詳細なデータの提供を求める場合があります。
Q4-19-2:(削除)
Q4-20:中間解析結果に基づき申請する場合、中間解析のためのカットオフ時点以降、申請時までに入手したデータも申請時に提出するデータに含めてもよいでしょうか。
A: 中間解析結果に基づき申請をする場合、提出する申請電子データに中間解析時のカットオフ時点までのデータは必ず含める必要がありますが、それに加えてカットオフ時点以降のデータを含めることも可能です。例えば、中間解析後一定期間が経過してから申請される場合等では、中間解析以降のデータを含めて申請することが有用と判断される場合があります。中間解析以降のデータを含めて申請する際には、カットオフ時点までのデータとそれ以降のデータのいずれかが明確になるようにし、当該データの取扱いについて、データガイドにおいて説明してください。
Q4-21:SDTMデータセット作成にSDTM IG v3.1.2 Amendment1を用いた場合の留意点はあるでしょうか。
A: PMDAにおいては、SDTM IG v3.1.2 Amendment1を適用してバリデーションを行うことができません。申請者側でも、SDTM IG v3.1.2 Amendment1以外のSDTM IGのバージョンを適用してバリデーションを実施した上で、必要な対応を行い、データセットの定義書にはバリデーション時に適用したバージョンを記載してください。なお、基本的には、SDTM-IG 3.1.3を用いてバリデーションを行うことが望ましいです。
Q4-22:SDTMデータセット、ADaMデータセットを格納するフォルダに、CDISC標準に準拠していないデータセットやデータセット以外のファイルを格納して提出することは可能でしょうか。
A: SDTMデータセット、ADaMデータセットを格納するフォルダには、CDISC標準に準拠したSAS XPORT形式のデータセットに加えて、データセットに付随するXML形式の定義書及びスタイルシート並びにPDF形式の文書等を格納し提出することは可能ですが、csvファイル、定義書以外のXML形式のファイル、CDISC標準に準拠していないSAS XPORT形式のデータセットを格納して提出することはできません。
Q4-22-1:Q4-22においてSDTMデータセット、ADaMデータセットを格納するフォルダに格納して提出することができないとされたファイルを提出する場合は、どのフォルダに格納すれば良いでしょうか。また、提出する際の留意点を教えてください。
A: 当該ファイルは、用途に応じて「misc」フォルダや「legacy」フォルダ等に格納して提出することが可能です。
例えば、ADaM作成に用いた参照データ(Lookup tables等の参照用テーブルやMetadata等)は、「misc」フォルダへ格納して提出することができます。また、ADaMデータセット作成用プログラムを提出し、当該ファイルがプログラム中で用いられている場合、プログラムに関連したファイルとして「programs」フォルダに格納することも可能です。
Multiple Imputation等による欠測値の補完に関連したデータ等も「misc」フォルダに格納して提出することができます。なお、この場合データセットの定義書及びデータガイド等において欠測値の取扱いについて説明してください。
CDISC標準以外の形式でまとめられたデータセットをCDISC標準の形式に変換して提出する際のトレーサビリティの説明として、CDISC標準への変換前のデータセット、トレーサビリティの説明とそれに付随するファイルは「legacy」フォルダに格納して提出することができます。
Q4-23:申請電子データ通知4(2)エに「単位についてはSI単位を使用することを原則とする。」とありますが、SI単位による値を格納するにあたり、PMDAが許容可能と考えるSI単位の範囲やその他の留意事項があれば示してください。
A: 現時点でPMDAは、国際度量衡局(BIPM:Bureau International des Poids et Mesures)が公表している報告書にある、SI単位の使用、及びSIとの併用が認められている非SI単位の併用、並びにSI接頭語の使用が、申請者が参考とすることのできる資料の1つと考えています。なお、接頭語は、原則として格納される数値が0.1から1000に含まれるように使用することとしてください。
Q4-24:中間解析が実施された臨床試験について、最終解析時のデータに加えて中間解析に関するデータの提出も必要とされる場合があるでしょうか。また、申請時に最終解析時のデータに加えて、中間解析に関するデータを併せて提出する場合の提出方法及び留意点を示してください。
A: 申請電子データの提出対象となる試験において、当該試験の中間解析の結果に基づき試験の中止/継続、重要な試験デザインの変更の要否や変更内容等に関する判断をした場合、申請時には最終解析時(もしくは申請の根拠となる最終のカットオフ時)のデータと併せて中間解析に用いた解析データセットの提出を求めることがあります。中間解析時の解析データセット及び解析用プログラムを提出する際は、解析データセットの定義書とともに「misc」フォルダに格納し、中間解析に用いた解析データセットを提出する旨をデータガイドで説明してください。また、中間解析の実施に際し中間解析用のSDTMを作成しない場合は中間解析時点のSDTMの提出は不要です。中間解析時のデータ提出の要否や、提出範囲等については、治験相談において相談してください。
Q4-25:申請電子データ通知3(3)に「長期投与試験の実施中に申請される場合、中間解析結果に基づき申請される場合等には、申請後に提出される当該臨床試験のデータは、既に提出されたデータに追加分のデータを含む形で提出すること。」とありますが、試験番号の異なる複数の試験間で継続している長期投与時のデータを集計する場合(例:第Ⅲ相試験(試験番号001)完了後、引き続き継続投与試験(試験番号002)に組み入れられた同一症例のデータを2試験間にわたり集計する場合。なお、第Ⅲ相試験のデータは申請時に提出済)、どのようにフォルダ名を規定してデータを追加提出すればよいでしょうか。
A: Q4-25のような状況で申請後にデータを追加提出する場合、追加提出データ(試験番号001と002の併合)は試験番号002のフォルダに格納して提出してください。なお、申請後のCDISC標準に準拠したデータのデータセットの追加提出方法及び手段についてはQ3-20に対する回答もあわせて参照してください。
複数試験間の併合データにおいては、データセット上でいずれの試験に由来するデータなのかがわかるようにしてください。また、データガイドにおいては、複数試験間の集計データを追加提出する旨とデータの集計方法、及び追加提出データにおける継続投与試験からの新規症例の組み入れの有無等をはじめとした留意点について説明してください。データの集計方法や提出方法の詳細については、必要に応じて医薬品申請電子データ提出方法相談において相談してください。
Q4-26:(削除)
Q4-27:同一試験内又は同一解析内において、複数のバージョンのCDISC標準、統制用語(以下「CDISC Controlled Terminology」という。)や外部辞書を用いた場合に留意する点はありますか。
A: 同一試験内又は同一解析内で使用するCDISC標準のバージョンについては、申請電子データ通知4(2)オにおいて、「同一臨床試験内及び同一解析内では統一したバージョンを用いること。同一臨床試験内又は同一解析内において他のバージョンを用いている部分がある場合には、データガイドにおいて、その使用の理由等とともに説明すること。」としているところです。ゲートウェイシステムでは、データセットの定義書に記載されたCDISC標準及びMedDRA、並びにゲートウェイシステムに登録されたCDISC Controlled Terminology等の単一バージョンを適用し、バリデーションが実施されます。したがって、申請者においてもデータセットの定義書に記載するSDTM IG、ADaM IG及びMedDRAのバージョン、並びにゲートウェイシステムに登録予定のCDISC Controlled Terminology等のバージョンを適用してバリデーションを実施し、必要なデータ修正又は説明をする必要があります。
Q4-28:データセット作成時に用いたCDISC Controlled Terminologyとは異なるバージョンのCDISC Controlled Terminologyを適用してバリデーションを実施しましたが、留意点はありますか。
A: 以下のように対応してください。
- 申請電子データ提出時の「試験データ提出」画面の「Terminology」欄にはバリデーションに適用したCDISC Controlled Terminologyのバージョンを記載してください。
- データガイドには、データセット作成とバリデーション実施に際して適用したCDISC Controlled Terminologyのバージョンが異なる旨、及びそれぞれのバージョンを記載してください。
Q4-29:医療用医薬品名データファイル(IDF)を用いて薬剤のコーディングを行い、Cross Reference Tool Japan(以下「CRT Japan」という。)を使用してWHODrug Globalデータに変換した上で、SDTMに格納する予定です。CRT Japanで変換された薬剤名等の情報以外は、それらの情報を格納する変数を空欄で提出することで差し支えないでしょうか。
A: CRT Japanで変換不可能な薬剤名等の情報がデータセットに含められるのであれば、CRT Japanで変換された薬剤名等の情報のみをSDTMに格納することで差し支えありません。
Q4-30:臨床試験実施時には日本語で有害事象名を収集しましたが、英語と日本語のSDTMデータセット(AEドメイン)を作成し、英語版の解析データセットに基づいて解析図表を作成しています。この場合、英語版のデータセットのみを提出することでも良いでしょうか。また、日本語版の解析データセットに基づいて解析図表を作成している場合はどうすべきでしょうか。
A: いずれの場合についても、英語版のデータセットで日本語版のデータセットの全ての情報が網羅されているのであれば、英語版のみを提出することで差し支えありません。なお、日本語版のデータセットを併せて提出することも可能です。
Q4-31:承認申請後に、承認申請時点で継続していた試験のデータを提出する際、eCTDのM5の総括報告書及び試験データは、それぞれ新規(new)のデータとして提出すべきでしょうか、それとも承認申請時点で提出したものを差換え(replace)にて提出すべきでしょうか。また、その際の格納方法について留意すべき点があれば教えてください。
A: 基本的には、eCTDのM5の総括報告書及び試験データについて、追加提出時に別途新規のデータとして、新たなフォルダ[study id / iss / ise]に格納することでも、既に提出されていたものを差し換えて既存のフォルダ[study id / iss / ise]に格納することでも提出可能です。しかしながら、いずれの方法がより望ましいかは、試験デザインや個別の試験における総括報告書及びデータの作成方法に依存する場合もあると考えられるため、提出方法については必要に応じて医薬品申請電子データ提出方法相談等で相談してください。
Q4-32:申請電子データ通知4(1)の「ただし、希少疾病用医薬品等について、令和2年4月1日より前に開始した試験については、その限りではない。」との記載及び申請電子データ通知Q&Aの問15に関連して、CDISC標準以外の形式の臨床試験の申請電子データを提出する場合の、提出内容及び提出方法を教えてください。また、CDISC標準以外の形式の申請電子データを提出する場合は、該当する試験や提出内容について、事前にPMDAに相談することと記載されていますが、対面助言においてCDISC標準以外の形式の申請電子データの提出内容を事前に相談する場合、どの相談で何を説明すればよいでしょうか。
A: CDISC標準以外の形式の臨床試験の申請電子データを提出する場合、少なくとも、CRF等により収集されたデータが格納された臨床試験データ(CDISC標準に準拠する場合のSDTMデータセットに相当する情報)、CTDに記載された解析結果を求めるための解析データセット、解析用プログラム及びデータセット定義書に相当するデータを提出する必要があります。そのため、CDISC標準以外の形式の申請電子データの提出内容を事前に相談する場合、医薬品申請電子データ提出免除相談を利用し、相談時には、「申請電子データに係る説明資料(Form B)」を用い、当該内容を説明するようにしてください。また、Annotated CRFやデータガイドに相当する情報の有無と提出の可否についても説明してください。なお、CDISC標準以外の臨床試験の申請電子データについては、技術的ガイド3.5に示すフォルダ構造のlegacyフォルダに格納してください。PK又はPK/PD解析に関する解析データセットについては、技術的ガイド3.5を参考に「cp」フォルダ等に格納することも可能です。
また、CDISC標準以外の形式の臨床試験の標準的な薬物動態解析については、「申請電子データに係る説明資料(Form B)」の「5.2臨床薬理領域の標準的な薬物動態解析又は薬力学解析」の項に記載してください。
Q4-32-1:申請電子データ通知4(1)に、「2(1)イ(イ)及び(ウ)の資料のうち、抗悪性腫瘍剤での第Ⅰ相試験以外の資料について、令和2年4月1日より前に開始した試験に限り、CDISC標準以外の形式のデータを提出することでも差し支えない。」と記載されていますが、これに該当する試験についてCDISC標準以外の形式の申請電子データを提出する場合の、提出内容及び提出方法を教えてください。
A: 当該記載に基づきCDISC標準以外の形式の臨床試験の申請電子データを提出する場合、少なくとも、CRF等により収集されたデータが格納された臨床試験データ(CDISC標準に準拠する場合のSDTMデータセットに相当する情報)、CTDに記載された解析結果を求めるための解析データセット、解析用プログラム及びデータセット定義書に相当するデータを提出する必要があります。また、Annotated CRFやデータガイドに相当する情報がある場合には併せて提出して下さい。これらのCDISC標準以外の臨床試験の申請電子データについては、基本的には技術的ガイド3.5に示すフォルダ構造のlegacyフォルダに格納してください。PK又はPK/PD解析に関する解析データセットについては、技術的ガイド3.5を参考に「cp」フォルダ等に格納することも可能です。
Q4-33:再審査時に提出が求められている製造販売後臨床試験の申請電子データについて、再審査申請に先立ち、PMDAによるバリデーション結果に従い適切に対応したデータを提出していたが、当該データの作成に用いた規格のバージョンが、再審査申請時にPMDAで受入れ可能な規格一覧のバージョンに含まれていなかった場合、当該データを再度修正して提出する必要がありますか。また、提出時に使用したバリデーションルールの受付が再審査申請時に終了していて選択できない場合、必要な対応はありますか。
A: ご質問のような場合、提出データ自体に変更がないのであれば、基本的には、特段の追加の対応は必要ありません。
Q4-34:申請電子データ通知2(1)イに「承認事項一部変更承認申請等において、過去の承認取得時に既に申請電子データを提出済みの臨床試験等について改めて提出する必要はない。」とありますが、先の申請において提出した臨床試験・解析に関する申請電子データと同一のデータを、先の申請の承認前に後の申請で用いたい場合、再度提出する必要はありますか。提出する必要がある場合、先の申請で提出したデータの作成に用いた規格のバージョンが、後の申請時にPMDAで受入れ可能な規格一覧のバージョンに含まれていなかった場合、同一のデータであっても再度修正して提出する必要がありますか。
A: 申請電子データ通知2(1)イに基づき、過去の承認申請において承認済みのデータと同一のデータを後の申請に用いる場合は、再度、同一のデータを提出する必要はありませんが、ご質問のように、後の申請の申請予定日が、先の申請の承認日より前であることが想定される場合は、先の申請において提出した臨床試験・解析に関する申請電子データと同一のデータであっても、後の申請において当該申請電子データを再度提出する必要があります。したがって、後の申請においても申請者において事前にバリデーションを実施し、必要な対応を行ってください。また、先の申請で提出したデータの作成に用いた規格のバージョンが、後の申請時にPMDAで受入れ可能な規格一覧のバージョンに含まれていなかった場合は、修正して提出する必要があります。
Q4-35:CDISC標準以外の形式のデータを提出する場合、薬剤コードにWHODrug Globalを使用する必要はありますか。
A: WHODrug Globalを使用する必要はありません。
Q4-36:申請電子データの提出対象となる臨床試験について、複数時点(カットオフ)の申請電子データを作成し、申請時に複数時点の申請電子データを提出する場合(例:中間解析時と最終解析時の2時点の申請電子データを提出する場合)、各申請電子データをどのフォルダに格納すればよいか教えてください。
A: 複数時点の申請電子データを提出する場合は、申請の根拠となる主要なカットオフ時点の全ての申請電子データを「m5¥datasets¥[study id / iss / ise]¥[analysis / tabulations]」フォルダに格納し、その他のカットオフ時点の申請電子データは「misc」フォルダに格納し提出することで差し支えありません。なお、主要なデータカットオフ時点の選択、その他のデータカットオフ時点に対応する申請電子データの提出要否及び提出範囲については、治験相談において相談してください。
主要なカットオフ時点の申請電子データには提出すべきデータを全て含める必要があることに留意してください。例えば、中間解析時と最終解析時(主要なカットオフに相当するとする)の2つの申請電子データを作成しており、中間解析時以降に更新のないデータセットが最終解析時の申請電子データに含まれていない場合、中間解析時以降に更新のないデータセットを最終解析時の申請電子データに追加して提出する必要があります。
臨床薬理領域の申請電子データについての質問
Q5-1:(削除)
Q5-2:標準的な薬物動態解析が実施された臨床試験とは、具体的にどのような臨床試験を指すでしょうか。
A: 「医薬品の臨床薬物動態試験について」(平成13年6月1日付け医薬審発第796号)において、標準的な薬物動態試験法と定義されている方法により薬物動態が評価された試験を指します。
Q5-3:申請電子データ通知Q&Aの問10について、「標準的な薬物動態解析が実施された臨床試験」における、「個々の臨床試験データ」とは、どのようなデータを指すでしょうか。
A: 薬物動態又は薬物動態/薬力学解析に関連するSDTMデータセット(例えば、PCドメインやPPドメイン)だけでなく、対象となる臨床試験の全てのSDTMデータセットを指します。
Q5-4:母集団解析が提出対象となる場合、母集団解析用のデータセットの作成に用いた個々の臨床試験のSDTMデータセット、標準的な薬物動態解析の解析データセット、並びに有効性及び安全性解析に関するデータセットも提出対象となるでしょうか。
A: 母集団解析については、母集団解析用のデータセット等、母集団解析に関する申請電子データのみが提出対象となります。なお、母集団解析用のデータセットの作成に用いた個々の臨床試験が、申請電子データ通知2(1)イに示されている申請電子データの提出の対象となる臨床試験に該当しない場合、当該試験のSDTMデータセット、標準的な薬物動態解析の解析データセット、並びに有効性及び安全性解析に関するデータセットの提出は必要ありません。
Q5-5:申請電子データ通知4(3)に「解析計画書に定めた除外理由以外の理由で解析から除外したデータ(例えば、解析時に外れ値であると判断して除外したデータ等)については、フラグにより特定できるようにする等、解析における取扱いが明確になるよう配慮すること。」とありますが、最終のデータセット(外れ値等を除外したもの)と、説明用として外れ値等を除外せず、外れ値等のデータにフラグをつけた全データの入った2種類のデータセットを提出することで、除外したデータの解析における取扱いを説明することは可能でしょうか。
A: 可能です。また、申請電子データ通知4(3)の当該記載は、解析から除外したデータが明確になる方法でデータを提出してほしいということを意図しており、その他の方法でも、意図に沿う方法であれば提出することが可能です。
Q5-6:(削除)
Q5-7:(削除)
Q5-8:(削除)
Q5-9:(削除)
Q5-10:(削除)
Q5-11:(削除)
Q5-12:(削除)
Q5-13:(削除)
Q5-14:(削除)
Q5-15:(削除)
Q5-16:(削除)
Q5-17:薬物動態又は薬物動態/薬力学に関する解析データセットに含むべき変数があれば教えてください。
A: いわゆる、Analysis readyなデータセットとするために必要な変数を含めてください。例えば、各被験者への投与量の情報、投与開始時点からサンプリング時点までの実際の経過時間の情報、PK指標として用いるAUC等の薬物動態パラメータ、PD指標として用いる測定値の変化量等の解析に必要な計算値、レコードを抽出するためのフラグ等が含まれている必要があります。なお、解析データセットをADaM形式に準拠して作成する場合には、ADaM IGで必須とされる変数も含める必要があります。
Q5-18:申請電子データ通知4(3)イ(イ)について、モデル構築過程をトレースできるように、共変量探索やモデル評価に用いたプログラムや出力ファイルを提出する必要があるのでしょうか。
A: 原則として基本モデル及び最終モデルが提出対象であり、共変量探索やモデル評価に用いたプログラムや出力ファイルは基本的には提出不要です。ただし、共変量探索やモデル評価のプロセスが複雑であり、これらのファイルを提出することが解析方法を理解する上で有用と申請者が考える場合等では、これらのファイルを提出することは可能です。
Q5-19:申請電子データ通知4(3)イ(イ)について、主要な結果が出力されたファイルを提出することとされていますが、主要な結果が解析報告書に記載されていた場合でも主要な結果が出力されたファイルを提出する必要がありますか。
A: 主要な結果が解析報告書に記載されている場合に、別途主要な結果が出力されたファイルを提出する必要はありません。
Q5-20:(削除)
Q5-20-1:(削除)
Q5-20-2:(削除)
Q5-21:薬物動態・臨床薬理領域の解析ソフトウェアでは、使用するバージョンにより解析結果に差異が生じますが、審査の過程で解析結果を確認される際には、申請者が解析に用いたバージョンのソフトウェアを用意されるのでしょうか。申請者側で最新のバージョンを用いて解析する必要があるのでしょうか。
A: PMDAで解析を実施する際に、可能な限り申請資料と同じ解析環境で検討することが望ましいと考えていますが、常に同一の解析環境を用意することは現実的でありませんので、解析環境により解析結果に差異が生じることは把握した上で、PMDAが申請者と異なるソフトウェアや異なるバージョンのソフトウェアを用いて解析する場合もあると考えています。また、必ずしも申請者側で最新のバージョンを用いて解析する必要はないと考えています。
Q5-22:申請時に提出した申請電子データについて、PMDAで不明な点がある場合や、PMDAの解析環境ではプログラムが動作しなかった場合は、申請後に照会事項として対応を求められることがありますか。
A: データやプログラムの内容に関する照会事項や問合せは原則として行わないと考えておりますので、データセット定義書や解析仕様書、プログラム手順書等の各種説明文書の内容の充実を図っていただきたいと考えています。
Q5-23:PMDAでプログラムの修正が必要であった場合には、プログラムを修正の上、再提出する必要がありますか。
A: いいえ。プログラムの再提出の必要はありません。
Q5-24:母集団解析(モデルに基づくシミュレーションを含む。)で検討に用いていない変数も提出を求められることがあるのでしょうか。
A: 申請後に新たな変数を加えたデータセットの作成と提出を求めることは考えていません。しかしながら、相談時には、例えば小児に対する用法・用量を検討するのであれば、体重や年齢の変数は加えてデータセットを作成する等、議論になる場合もあると考えています。
Q5-25:(削除)
Q5-26:同一試験内の複数時点(カットオフ)でPK解析を実施した場合、各時点のADaMデータセットはどのフォルダに格納すればよいか教えてください。
A: 例えば、有効性・安全性の解析が1回実施される期間中にPK解析を複数回実施される場合やPKデータ取得のタイミング等の理由により、有効性・安全性の解析とPK解析のタイミングが異なる場合等には、ADaMデータセットのファイル名を解析時期によって変える等して作成して、
[study id / iss / ise]¥analysis¥adam¥datasetsフォルダに格納することで差し支えありません。上記の方法で対応できない場合や試験全体で複数のカットオフで解析を実施する場合(例:中間解析と最終解析を実施して提出するような場合)には、評価上最も重要と考えられる解析時点のADaMデータセットは[study id / iss / ise]¥analysis¥adam¥datasetsフォルダに格納し、その他のADaMデータセットは「misc」フォルダに格納してください。評価上最も重要と考えられる解析時点についての相談や、提出の要否等については、治験相談において相談してください。また、データの格納方法に迷う場合には、医薬品申請電子データ提出方法相談において相談してください。
Q5-27:第Ⅰ相試験、臨床薬理試験等の成績及び臨床薬理領域の解析に関し、CDISC標準以外の形式で作成した解析データセットに併せて提出するデータセット定義書は、決まった様式がありますか。また、データセット定義書は英語でもよいでしょうか。
A: 決まった様式はありません。申請者が作成したデータセット定義書をそのまま提出することで差し支えなく、データセット定義書は英語でも構いません。
Q5-28:技術的ガイド4.2.2.1に、解析に関する詳細情報が、解析データセット自体に含まれる場合には、その旨を明示することで差し支えない旨の記載がありますが、当該事項を記載することが推奨される文書があれば教えてください。
A: 提出をお願いしている解析仕様書等、申請時に提出する文書中に記載してください。なお、提出する解析計画書等に当該解析情報が含まれる場合にも、その旨を解析仕様書等に記載してください。
Q5-29:技術的ガイド、別紙5のプログラム手順書を英語で提出することは可能ですか。
A: 可能です。
Q5-30:標準的な薬物動態解析を実施した臨床試験について申請電子データを提出する場合、臨床薬理領域の解析に関するAnalysis Results Metadataの提出対象となる解析の例を示してください。
A: 標準的な薬物動態解析が実施された臨床試験について、ノンコンパートメント解析により薬物動態又は薬力学パラメータを算出する解析は、Analysis Results Metadataの提出対象となりません。一方、解析データセットの提出対象となる、薬物動態又は薬力学パラメータの統計学的な検討に用いた解析は、Analysis Results Metadataの提出が推奨されます。ただし、解析データセットをADaM以外の形式で提出する場合は、Analysis Results Metadataの提出は不要です。
Q5-31:申請電子データ通知Q&A問18において、日本人と外国人の双方に対して実施された第Ⅰ相試験及び申請電子データ通知2(1)イ(ウ)の資料のうち標準的な薬物動態解析が実施された臨床試験について、必ずしも有効性及び安全性に関する解析データセットの提出を要しない場合があると記載されています。これらの試験の安全性評価に関して、臨床試験データ(SDTM等)を用いて、有害事象や臨床検査値データの単純な集計のみ実施されており、解析データセットの形式(ADaM等)に関わらず解析データセットが作成されていない場合、解析データセットを作成し提出することが必要となりますか。
A: これらの試験の安全性評価に関しては、臨床試験データ(SDTM等)を用いて、有害事象や臨床検査値データの単純な集計のみが実施されており、解析データセットが作成されていない場合は、解析データセットを提出する必要はありません。
Q5-32:技術的ガイド4.2.2.1に関連して、薬物動態又は薬物動態/薬力学に関する解析仕様書に準じて提出すべき情報を含む資料として、Phoenix Projects (*.phxproj)のText OutputのCore outputファイル又は当該ファイル情報を含むPhoenix Projects (*.phxproj)を提出することは可能でしょうか。
A: 可能です。
Q5-33:薬物相互作用の検討を目的とした生理学的薬物速度論モデル解析に関する提出ファイルとして、解析報告書中で、申請薬物以外の薬物(以下「薬物A」とする。)の生理学的薬物速度論モデルを用いた解析を実施している場合、薬物Aの生理学的薬物速度論モデルに関するファイルも提出する必要はありますか。
A: はい、提出する必要があります。
Q5-34:申請電子データ通知Q&A問17(2)②として、「申請者が検証的試験の用法・用量の設定根拠と考える母集団解析」が申請電子データの提出対象として例示されていますが、検証的試験の用法・用量の設定根拠として用いられた母集団解析について、検証的試験実施後に検証的試験で得られたデータを用いて解析データセット及び/又はモデルが更新され、更新後の解析が申請用法・用量の設定根拠として用いられた場合に、更新後の解析に関する申請電子データのみを提出し、更新前の解析に関する申請電子データを提出しないことは可能ですか。
A: 可能です。
参照先
過去に発出した申請電子データに関する通知については、「申請電子データ提出」の「関連通知・様式等」を参照ください。
- 申請電子データ通知
令和6年4月8日付け医薬薬審発0408第3号厚生労働省医薬局医薬品審査管理課長通知 「承認申請時等の電子データ提出に関する取扱いについて」の一部改正について[429KB] - 申請電子データ通知Q&A
令和6年4月8日付け厚生労働省医薬局医薬品審査管理課事務連絡 「承認申請時等の電子データ提出に関する取扱いについて」に関する質疑応答集(Q&A)について[311KB] - ゲートウェイ申請通知
令和4年4月1日付け薬生薬審発0401第7号厚生労働省医薬・生活衛生局医薬品審査管理課長通知 「ゲートウェイシステムを利用した新医薬品の承認申請等について[193KB] - 技術的ガイド
令和6年4月8日付け薬機審長発第1345号、薬機RS長発第21号独立行政法人医薬品医療機器総合機構審査センター長、RSセンター長連名通知 「承認申請時等の電子データ提出に関する技術的ガイドについて」の一部改正について[506KB] - 申請・届出受付等業務実施要綱
令和4年12月26日付け薬機発第1226041号独立行政法人医薬品医療機器総合機構理事長通知「独立行政法人医薬品医療機器総合機構が行う審査等業務に係る申請・届出等の受付業務の取扱いについて」[399KB] - 新医薬品承認審査予定事前面談実施要綱
平成26年10月6日付け薬機発第1006001号独立行政法人医薬品療機器総合構理事長通知「新医薬品承認審査予定事前面談実施要綱について」[419KB] - 対面助言、証明確認調査等の実施要綱通知
平成24年3月2日付け薬機発第0302070号独立行政法人医薬品医療機器総合機構理事長通知「独立行政法人医薬品医療機器総合機構が行う対面助言、証明確認調査等の実施要綱等について」[2.40MB] - 申請電子データに係る説明資料(Form A)
「申請電子データに係る説明資料(Form A)の作成要領」[89.4KB]
「申請電子データに係る説明資料(Form A)」の様式[86.1KB] - 申請電子データに係る説明資料(Form B)
「申請電子データに係る説明資料(Form B)の作成要領」[69.5KB]
「申請電子データに係る説明資料(Form B)」の様式[67.1KB] - 医薬品の臨床薬物動態試験について
平成13年6月1日付け 医薬審発第796号厚生労働省医薬局審査管理課長通知「医薬品の臨床薬物動態試験について」[344KB]
申請電子データに関するFAQの改訂箇所について
- 申請電子データに関するFAQ(令和7年3月31日公開)の改訂箇所について[303KB]
- 申請電子データに関するFAQ(令和6年4月8日公開)の改訂箇所について[518KB]
- 申請電子データに関するFAQ(令和5年10月2日公開)の改訂箇所について[319KB]
- 申請電子データに関するFAQ(令和5年4月3日公開)の改訂箇所について[195KB]
- 申請電子データに関するFAQ(令和4年6月27日公開)の改訂箇所について[259KB]
- 申請電子データに関するFAQ(令和4年4月1日公開)の改訂箇所について[769KB]
- 申請電子データに関するFAQ(令和3年4月1日公開)の改訂箇所について[607KB]
- 申請電子データに関するFAQ(令和2 年3月23日公開)の改訂箇所について[64.2KB]
- 申請電子データに関するFAQ(平成31年4月10日公開)の改訂箇所について[218KB]
- 申請電子データに関するFAQ(平成31年1月24日公開)の改訂箇所について[144KB]
- 申請電子データに関するFAQ(平成30年5月17日公開)の改訂箇所について[91.1KB]
- 申請電子データに関するFAQ(平成30年3月7日公開)の改訂箇所について[150KB]
- 申請電子データに関するFAQ(平成29年9月15日公開)の改訂箇所について[123KB]
- 申請電子データに関するFAQ(平成29年2月3日公開)の改訂箇所について[251KB]
- 申請電子データに関するFAQ(平成28年8月29日公開)の改訂箇所について[97.2KB]
- 申請電子データに関するFAQ(平成28年7月8日公開)の改訂箇所について[154KB]