
PMDAでは、医薬品、医療機器、再生医療等製品等の品質、有効性、安全性について現在の科学技術水準に基づき承認審査を行っています。承認審査のほか審査関連業務は、承認申請資料などに関する相談を受ける「相談業務」、申請資料の倫理的・科学的信頼性を調査する「信頼性調査」、製品の製造体制を調査する「GMP/QMS/GCTP調査」など多岐にわたります。
PMDAは、医薬品、医療機器、再生医療等製品等の開発の初期から製造販売後にかけて、様々な業務を行っています。それぞれの業務の詳細については、各業務のページをご覧ください。
医薬品、医療機器、再生医療等製品の開発から市場に出るまでの流れとPMDAの関わる業務
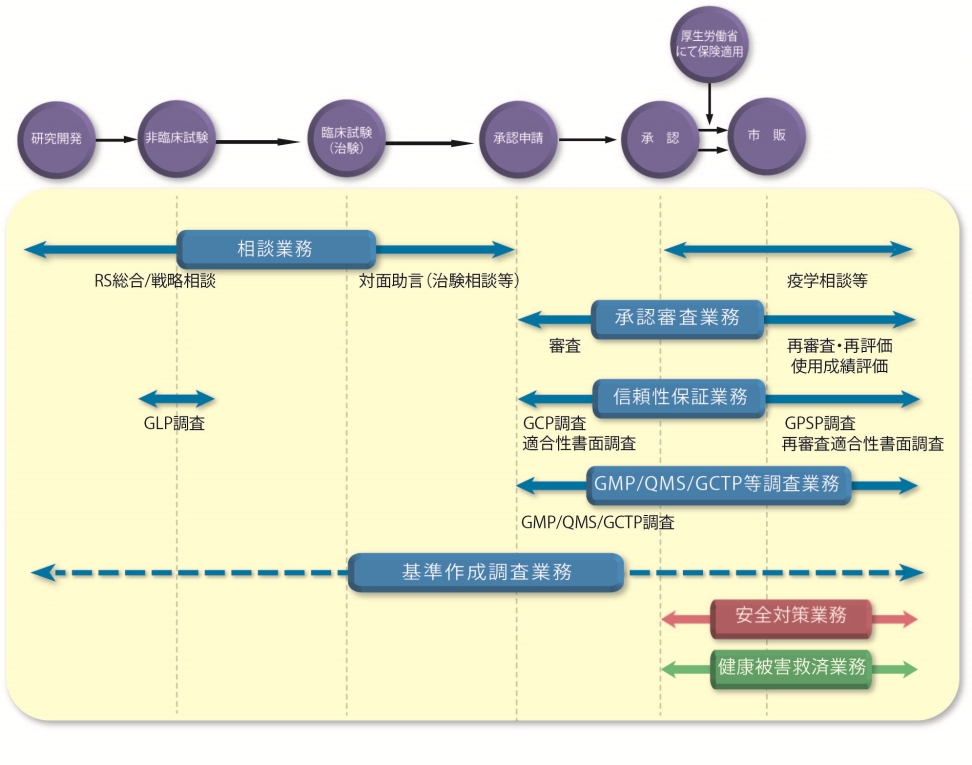
相談業務
PMDAでは、医薬品、医療機器、再生医療等製品の治験や申請資料に対し、指導・助言を行っています。この他、医薬部外品などの簡易相談についても、申込者に対して指導・助言を行っています。また、2011年(平成23年)7月からは、日本発の革新的医薬品、医療機器、再生医療等製品の創出に向けて、有望なシーズを持つ大学・医療機関、ベンチャー企業を主な対象とした薬事戦略相談を実施してきました。2017年(平成29年)4月からは、相談事業の一層の充実を図るため、薬事戦略相談事業として実施している面談のうち、事前面談及び本相談については、名称を「レギュラトリーサイエンス戦略相談(RS戦略相談)」事業と改称して引き続き実施するとともに、個別面談については、対象を拡大し、新たに「レギュラトリーサイエンス総合相談(RS総合相談)」事業として実施しています。
治験関連業務
治験の実施にあたって、治験依頼者(製造販売業者等)及び自ら治験を実施する者(医師、歯科医師等)は、治験計画の届出及び治験中の副作用、不具合等の報告が義務づけられています。PMDAでは、治験計画届及び治験中の副作用、不具合等報告の受付等を行っています。また、治験審査委員会(IRB)についても、各医療機関からPMDAに登録された情報を公表しています。
承認審査業務
医薬品等承認審査業務
医薬品の承認審査では、薬学、医学、獣医学、理学、生物統計学などの専門知識を有する審査員が、「品質」「薬理」「薬物動態」「毒性」「臨床」「生物統計」を担当し、審査チームを形成して審査を行います。また、審査の過程では、外部専門家との意見交換(専門協議)を行い、より専門性の高い見地から審査することを目指しています。
また、より優れた医薬品をより早く医療現場に提供するため、審査期間の目標を設定し、業務の迅速化に取り組んでいます。
さらに、日本、アメリカ、ヨーロッパにおける新医薬品の承認審査資料関連規制の整合化を図ることにより、データの国際的な相互受け入れを実現することを目的とした医薬品規制調和国際会議(ICH)に参加するとともに、同会議で合意された内容を積極的に承認審査に取り入れています。
医薬品等の承認審査では、「新医薬品」のほかに、「後発医療用医薬品(すでに承認されている医薬品と同一性が認められる医薬品)」、薬局・薬店で医師の処方せんなしに購入できる「一般用医薬品(OTC)・要指導医薬品」、その他「医薬部外品」を審査しています。
また、医薬品の再審査・再評価や、遺伝子組換え生物、再生医療(細胞組織利用医薬品)、遺伝子治療用医薬品などの確認申請の審査なども行っています。
新医薬品承認審査実務に関わる審査員のための留意事項[239KB]
新医薬品の承認審査の進捗状況の確認について[158KB]
新医薬品の承認審査の進捗状況の確認について(2016年(平成28年)10月3日一部改正)[121KB]
製造販売後調査等の実施計画の策定に関する検討の進め方について[221KB]
医薬品の製造販売後調査等の実施計画の策定に関する検討の進め方について(2019年(平成31年)3月14日付け薬生薬審発0314第4号、薬生安発0314第4号)[315KB]
独立行政法人医薬品医療機器総合機構審査等業務及び安全対策業務に係る不服等の対応について[10.6KB]
医薬品等の承認審査における競合品目選定の基本的考え方について(2025年(令和7年)4月3日)[95.1KB]
医療機器承認審査業務
医療機器は、医薬品と同じく、疾病の診断、治療、予防など、医療に用いる製品という特性と、メスやピンセットから、MRI、ペースメーカーまで、製品ごとに基になる技術・素材が異なり、使用形態、リスクの程度など、多種多様な製品に応じて合理的な規制が必要であるという特性があります。
PMDAでは、これらの医療機器のうちハイリスク医療機器(例:人工心臓、ペースメーカー、冠動脈ステント、人工血管、人工関節、人工腎臓等)を中心に、承認審査を行っています。
医療機器の承認審査では、このような医療機器の特性を踏まえた上で、より優れた医療機器をより早く医療現場に提供するため、審査期間目標を設定し、業務の迅速化に取り組んでいます。
実際の承認審査にあたっては、医用工学、生体工学、バイオマテリアルなどについて詳しい知識を持つ工学系の審査員の他、医学、歯学、薬学、獣医学、理学、生物統計学などの専門知識を有する審査員が非臨床、臨床、生物統計を担当し、複数名で審査を行います。また、審査の過程で、外部専門家との意見交換(専門協議)を行い、より効率的、専門的な審査を目指しています。
さらに、国際的にみて整合性のとれた医療機器の審査体制を整えるため、国際医療機器規制当局フォーラム(IMDRF)に参加するとともに、同会議で合意された内容を積極的に取り入れ、国際標準化機構(ISO)、国際電気標準会議(IEC)などの規格を取り入れた承認審査体制を構築しています。
なお、管理医療機器であって、認証基準が策定されたものは第三認証制度に移行しています。また、一般医療機器についてはPMDAへ届け出ることとなっています。
再生医療等製品承認審査業務
2013年(平成25年)11月27日に公布された医薬品医療機器法において、再生医療等製品が新たに定義されました。再生医療等製品は、人や動物の生きた細胞・組織を用いた製品や遺伝子治療用の製品であることから、従来の医薬品・医療機器と異なる性質を持っています。
例えば、生きた細胞を用い、製品の品質が不均一となる場合は、有効性が推定され、安全性が確認されれば、条件及び期限付きで特別に早期に承認できる仕組みとして、「条件及び期限付き承認制度」が導入されました。
信頼性保証業務(GLP/GCP/GPSP)
信頼性保証業務とは、医薬品、医療機器又は再生医療等製品の承認申請又は再審査・再評価/使用成績評価申請された品目について、申請書に添付された資料(承認申請資料又は再審査・再評価/使用成績評価申請資料)が、厚生労働大臣の定める基準である医薬品、医療機器又は再生医療等製品GLP(安全性に関する非臨床試験の実施の基準に関する省令に示された基準)、医薬品、医療機器又は再生医療等製品GCP(臨床試験の実施の基準に関する省令に示された基準)、医薬品、医療機器又は再生医療等製品GPSP(製造販売後の調査と試験の実施の基準に関する省令に示された基準)及び「申請資料の信頼性の基準(医薬品医療機器法施行規則第43条、第61条、第114条の22、第114条の42、第137条の25又は第137条の42)」に従って収集され、かつ、作成されたものであるかについて調査することです。PMDAでは、当該申請資料がGLP、GCP及びGPSPに従って倫理的、科学的に適切に実施された試験の成績に基づいているかどうか、また、「申請資料の信頼性の基準」に従って、試験結果に基づいて適切かつ正確に作成されているかどうかを実地及び書面で調査を行います。
GMP/QMS/GCTP適合性調査業務
GMP/QMS/GCTP適合性調査とは、医薬品、医薬部外品、医療機器、再生医療等製品を製造している製造所が適正な管理の下にこれら医薬品等を製造しているかどうかを調査するものです。
PMDAでは、生物学的製剤等を製造しているこれら製造所に対して、GMP省令(医薬品及び医薬部外品の製造管理及び品質管理 の基準に関する省令)、QMS省令(医療機器及び体外診断用医薬品の製造管理及び品質管理の基準に関する省令)、GCTP省令(再生医療等製品の製造管理及び品質管理の基準)に基づいてそれら製品が適切 に製造されているかどうかを調査しています。この調査は製造所に赴き実地に調査するほか、書面による調査を行います。
再審査・再評価業務
PMDAでは、医薬品の再審査及び再評価、医療機器の使用成績評価に関する業務を行っています。
登録認証機関に対する調査等業務
医療機器及び体外診断用医薬品のうち、厚生労働大臣が基準を定めて指定したものを製造販売しようとする際には、登録認証機関の認証を受けなければならないこととなっています。PMDAはこの認証機関の登録や登録の更新にあたり、登録認証機関(登録認証機関になろうとする者を含む)が登録の基準に適合しているかどうかについて必要な調査を行います。